
文章链接
Avsec, Ž., Agarwal, V., Visentin, D., Ledsam, J. R., Grabska-Barwinska, A., Taylor, K. R., Assael, Y., Jumper, J., Kohli, P., & Kelley, D. R. (2021). Effective gene expression prediction from sequence by integrating long-range interactions. Nature Methods, 18, 1196–1203. https://doi.org/10.1038/s41592-021-01252-x
简介
Enformer:一个用于预测基因表达的深度学习架构。
摘要
非编码DNA如何在不同细胞类型中决定基因表达是一个重大的未解之谜,而在人类遗传学中的许多重要下游应用都依赖于对这个问题的深入理解。在此,我们报告通过使用一种称为Enformer的深度学习架构,显著提高了从DNA序列预测基因表达的准确性。该架构能够整合基因组中的长程相互作用信息(距离可达100kb)。这种改进使得对自然遗传变异和通过大规模平行报告基因检测(massively parallel reporter assays)测量的饱和突变对基因表达的影响预测更加准确。此外,Enformer学会直接从DNA序列预测增强子-启动子相互作用,其准确度可与直接使用实验数据作为输入的方法相媲美。我们预计这些进展将有助于更有效地精确定位人类疾病的相关性,并为解释顺式调控元件(cis-regulatory)的进化提供一个理论框架。
创新点
-
使用
transformer架构替代传统的卷积神经网络,能够整合长距离(最远100kb)的DNA序列信息。这比之前的模型(如Basenji2)的20kb范围更大。 -
在基因表达预测方面取得显著提升:
-
皮尔逊相关系数从0.81提升至0.85
-
在不同组织和细胞类型的预测准确度都有提高
-
-
模型可以有效识别和使用调控元件:
-
能够识别增强子及其与启动子的相互作用
-
能够识别绝缘子元件和拓扑相关结构域(TAD)边界
-
transformer架构可参考这篇文章:
主要内容
读前须知
- 论文解读尽可能的还原原文,若有不恰当之处,还请见谅;
- 排版上,插图会尽量贴近出处,而扩展图之类的,会放置末尾处;
- 左边👈有目录,可自行跳转至想看的部分;
- 部分专业术语翻译成中文可能不太恰当,此时会用括号标明它的英文原文,如感受野(
Receptive field)。请注意,仅首次出现会标明;
背景
从DNA序列预测基因表达和染色质状态的模型,有望更好地理解转录调控以及它如何受到与人类疾病和性状相关的众多非编码遗传变异的影响。这些模型补充了基于人群的关联研究,后者通常仅限于常见变异,并且由于连锁不平衡(LD)而难以区分因果关系和关联性。此外,人类遗传变异的实验验证工作量大,且仅限于可在实验室中重现的细胞类型或组织,这使得在相关生物环境中测试所有感兴趣的变异变得不可行。虽然基于序列的计算模型原则上可以克服这些挑战,但它们的准确性仍然有限,使得从序列预测基因表达成为一个关键的未解决问题。
连锁不平衡:基因组中相邻位置的遗传变异倾向于一起遗传的现象。即某些等位基因或遗传变异在群体中共同出现的频率高于随机预期。
深度卷积神经网络(CNNs)在预测人类和小鼠基因组的基因表达方面达到了目前的最高水平。然而,这些模型在做出预测时,只能考虑距转录起始位点(TSS)最远20kb的序列元件,因为卷积的局部性限制了网络中远距离元件之间的信息流动。许多经过深入研究的调控元件,包括增强子、抑制子和绝缘子,都可以从超过20kb的距离影响基因表达。因此,增加远距离元件之间的信息流动是提高预测准确性的一个有希望的途径。
增强子(Enhancer):可以增强基因转录的DNA序列;
抑制子(Repressor):能够降低或抑制基因表达的DNA序列;
绝缘子(Insulator):位于拓扑相关结构域(TAD)的边界,主要功能是阻隔/隔离不同调控区域之间的相互作用,防止增强子错误调控非目标基因;这三种元件共同参与精确调控基因表达,形成复杂的调控网络。
核心内容
在这项工作中,我们引入了一种基于自注意力(self-attention)的神经网络架构来实现这一目标。我们将机器学习问题定义为在多任务设置下预测长DNA序列中的数千个表观遗传和转录数据集。在人类和小鼠基因组的大部分区域进行训练,并在保留的序列上进行测试,我们观察到相对于之前没有自注意力的最先进模型,预测与测量数据之间的相关性有所提高。我们证明了对长程信息的更有效利用,这一点通过CRISPRi增强子检测得到了验证。该模型还能产生更准确的突变效应预测,这一点通过直接突变检测和人群eQTL研究得到了验证。
自注意力机制:允许模型在处理序列时,让每个位置都能"关注"并整合来自所有其他位置的信息。在Enformer中,这使得模型能够将位于不同位置的调控元件(如启动子和增强子)的信息关联起来。
eQTL:表达数量性状位点,指能影响基因表达水平的DNA序列变异。这些变异可以位于基因内部或调控区域,通过影响转录调控或其他机制来改变基因的表达量。
CRISPRi增强子检测:利用CRISPR干扰(CRISPR interference, CRISPRi)技术研究增强子功能的一种方法。CRISPRi通过抑制特定基因组区域的活性,可以系统性地筛选和验证增强子对基因表达的调控作用。
成果
Enformer 改进了基因表达预测
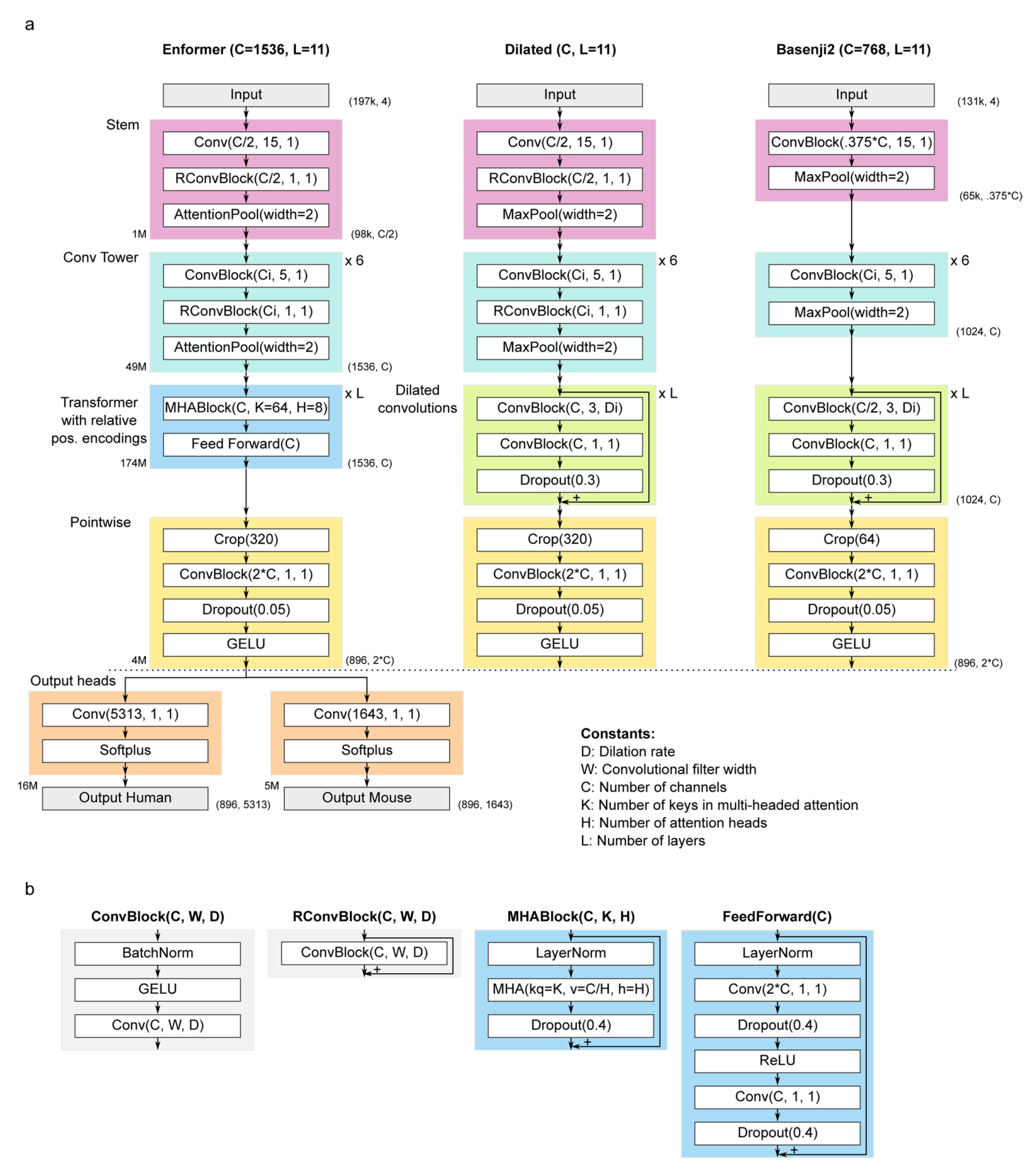
我们开发了一个名为Enformer(enhancer和transformer的组合词)的新模型架构,用于预测人类和小鼠的基因表达和染色质状态(图1a和扩展数据图1)。
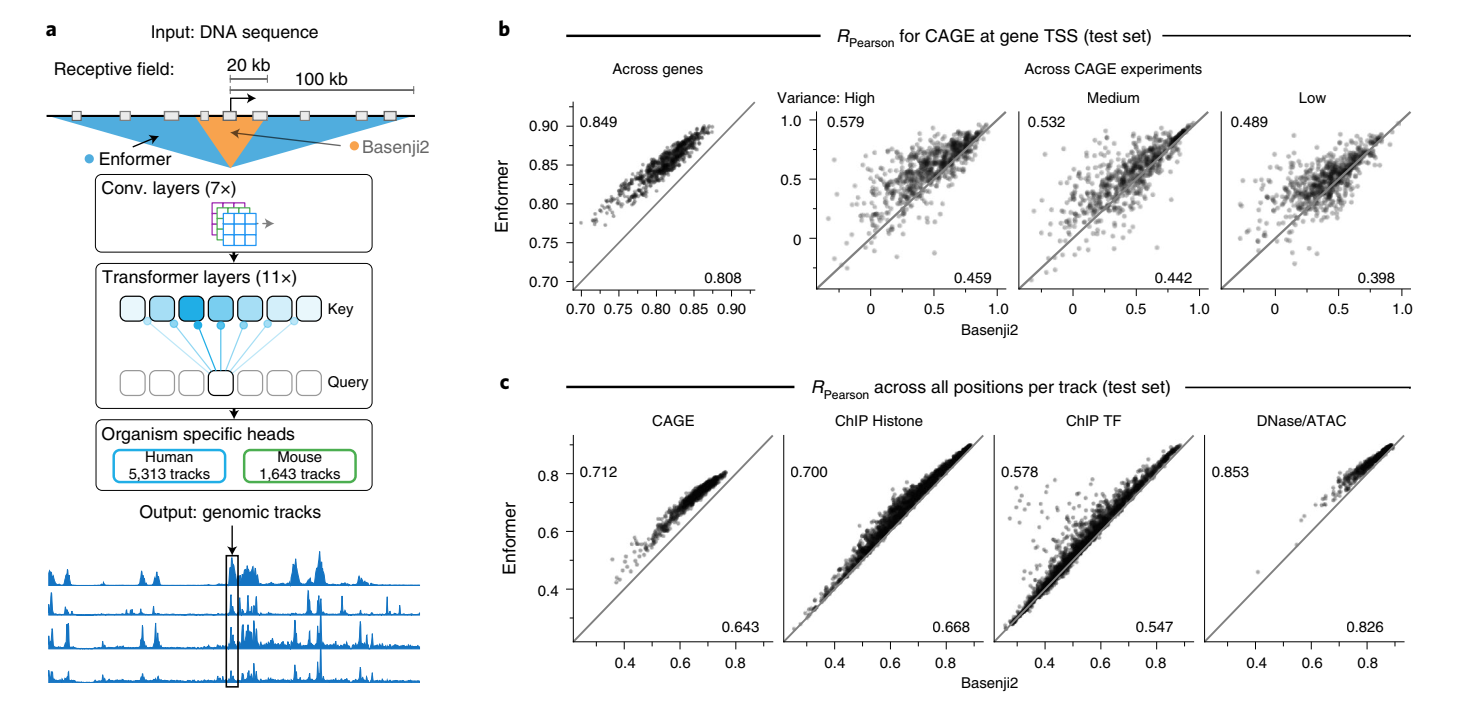
Transformer是一类在自然语言处理(NLP)中取得重大突破的深度学习模型,最近也被应用于建模短DNA序列。它们由注意力层组成,这些层通过计算所有其他位置表征的加权和来转换输入序列中的每个位置。注意力权重取决于它们当前表征向量的嵌入和位置之间的距离。这使得模型能够,例如,通过收集所有相关区域(如调控该基因的增强子)的信息来完善在转录起始位点的预测。由于每个位置直接关注序列中的所有其他位置,它们能够实现远距离元件之间更好的信息流动。相比之下,卷积层由于其局部感受野,需要许多连续层才能到达远距离元件。使用transformer层使我们能够显著增加感受野,从而接触到距离达100kb的远距离调控元件,而之前最先进的模型Basenji2或ExPecto只能达到20kb(扩展数据图1)。这种感受野的增加很重要,因为它大大扩展了模型可见的相关增强子数量,从<20kb的47%增加到<100kb的84%(根据高置信度增强子-基因对的比例估计)。
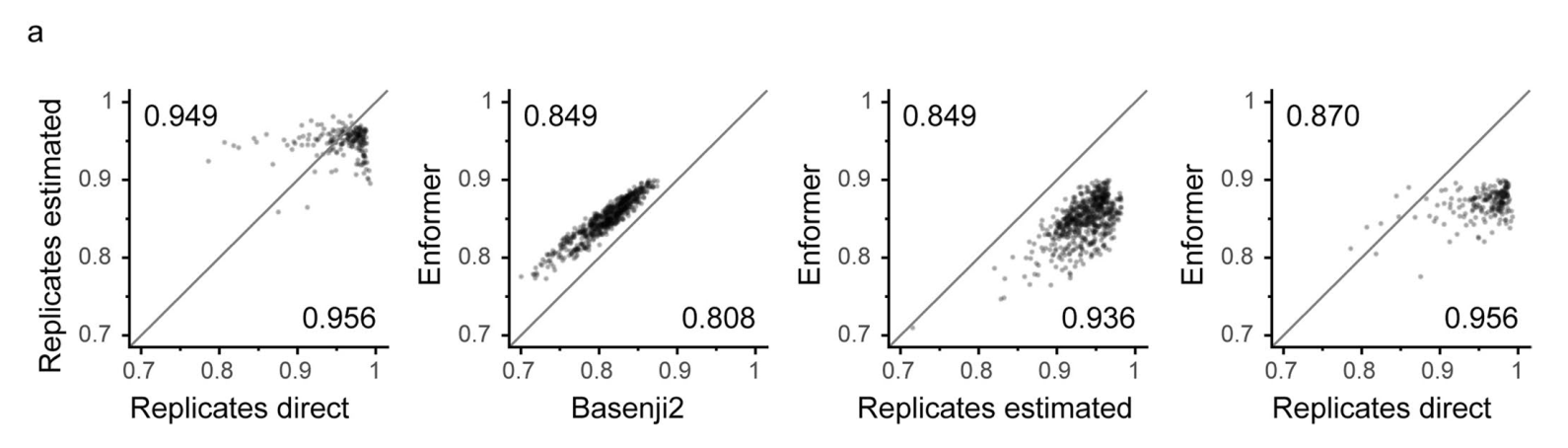
Enformer在预测人类蛋白质编码基因转录起始位点的RNA表达(通过CAGE测量)方面显著优于之前最好的模型Basenji2,平均相关性从0.81提高到0.85(图1b左)。这种性能提升是Basenji1和Basenji2之间性能提升的两倍,并缩小了三分之一到实验水平精确度(估计为0.94)的差距(扩展数据图2)。基因表达预测也更好地捕捉到了组织或细胞类型特异性(图1b右),包括密切相关的样本(扩展数据图3)。这种性能改进在所有四种基因组广泛轨迹类型中都是一致的,包括在各种细胞类型和组织中测量转录活性的CAGE、组蛋白修饰、转录因子结合和DNA可及性(图1c)。对CAGE的性能改进最大,这可能是因为组织特异性基因表达强烈依赖于远距离元件。
测量转录活性的CAGE:CAGE(Cap Analysis of Gene Expression)是一种测量转录起始活性的方法。它通过捕获mRNA 5’端的帽结构(cap),结合高通量测序,能够精确定位转录起始位点(TSS),并定量转录本的表达水平。这种技术特别适合分析启动子活性、TSS使用的变化,以及基因表达调控。
组蛋白修饰:发生在组蛋白特定氨基酸残基上的共价化学修饰,这些修饰通过改变染色质的包装状态(紧致或松散),影响转录因子的结合,从而动态调控基因的活性。
转录因子结合:转录因子(Transcription Factors, TFs)与DNA特定序列(通常是启动子或增强子上的顺式作用元件)的结合过程,这是基因表达调控的关键步骤。
DNA可及性:DNA在染色质中的暴露程度,它决定了转录因子、RNA聚合酶等调控蛋白能否结合到特定的DNA区域。DNA可及性是基因表达调控的关键因素,受染色质结构的动态调节影响。
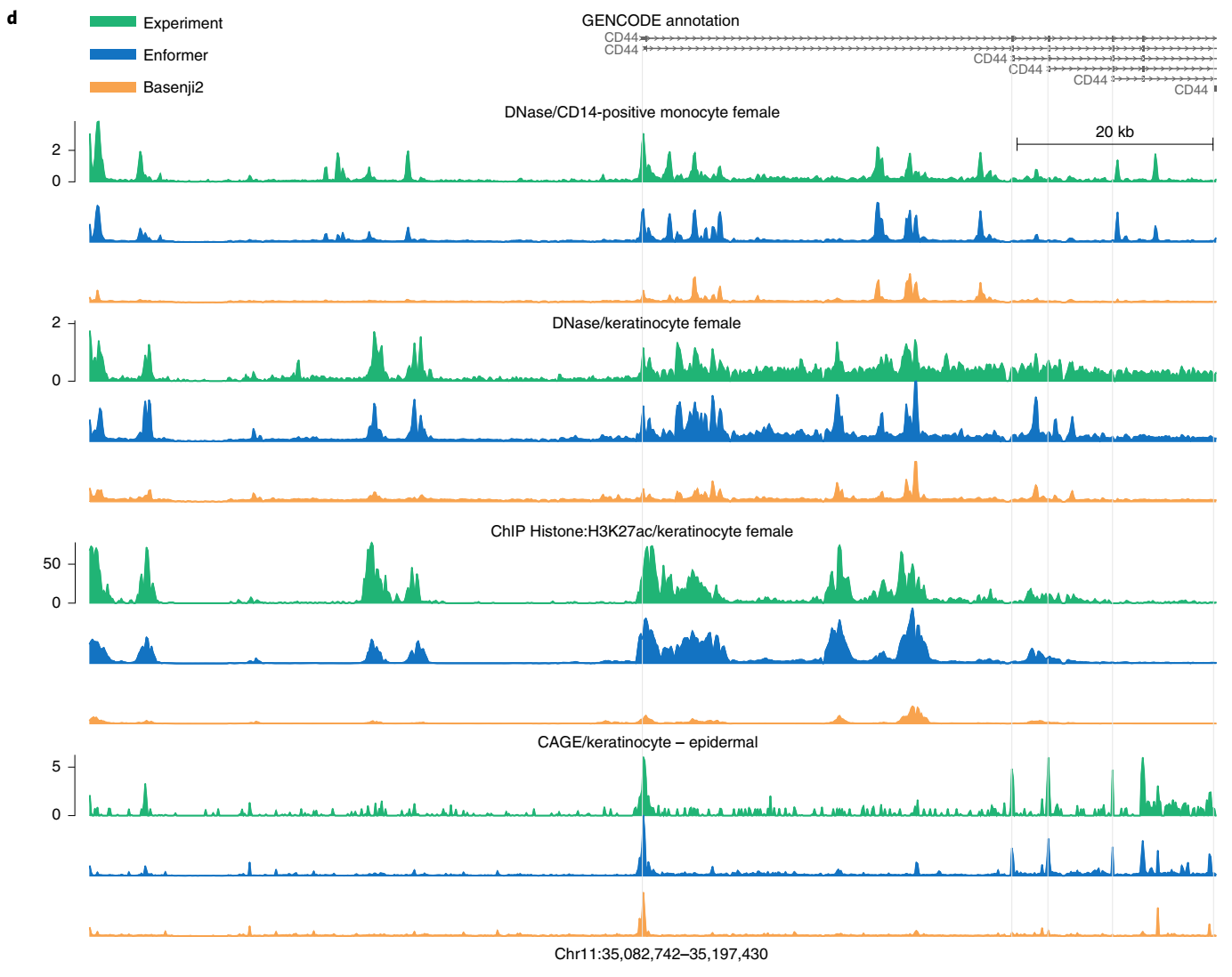
在可视化基因组的观察和预测轨迹时,预测准确性的提高也是显而易见的(图1d)。Enformer在预测由RNA-seq测量的基因表达水平方面也优于ExPecto,在跨基因(Spearman r为0.850对0.812)和跨组织(Spearman r为0.451对0.368)评估中都表现更好(扩展数据图4)。这些结果证实Enformer架构在从DNA序列预测表观遗传标记和基因表达方面都提高了预测准确性。
为了确定相比于Basenji2中使用的扩张卷积,注意力层的具体优势,我们用扩张卷积替换了注意力层,并调整了学习率以获得最佳性能。注意力层在所有模型大小、层数和训练数据点数量上都优于扩张卷积(扩展数据图5a)。更大的感受野确实至关重要,因为当我们通过用局部注意力层替换全局注意力层,将Enformer的感受野限制为与Basenji2相同时,我们观察到性能显著下降(扩展数据图5b)。我们注意到,增加参数数量改善了模型性能,这与自然语言处理领域的最新进展一致。
Enformer在transformer层中使用自定义的相对位置基函数,以更容易区分近端和远端调控元件,并区分转录起始位点上游和下游的位置。这两个特性相比于自然语言处理文献中通常使用的相对基函数和绝对位置编码都提供了明显的性能改进(扩展数据图6a、b)。总的来说,这些结果证实注意力层比扩张卷积更适合基因表达预测。
Enformer 关注细胞类型特异性增强子
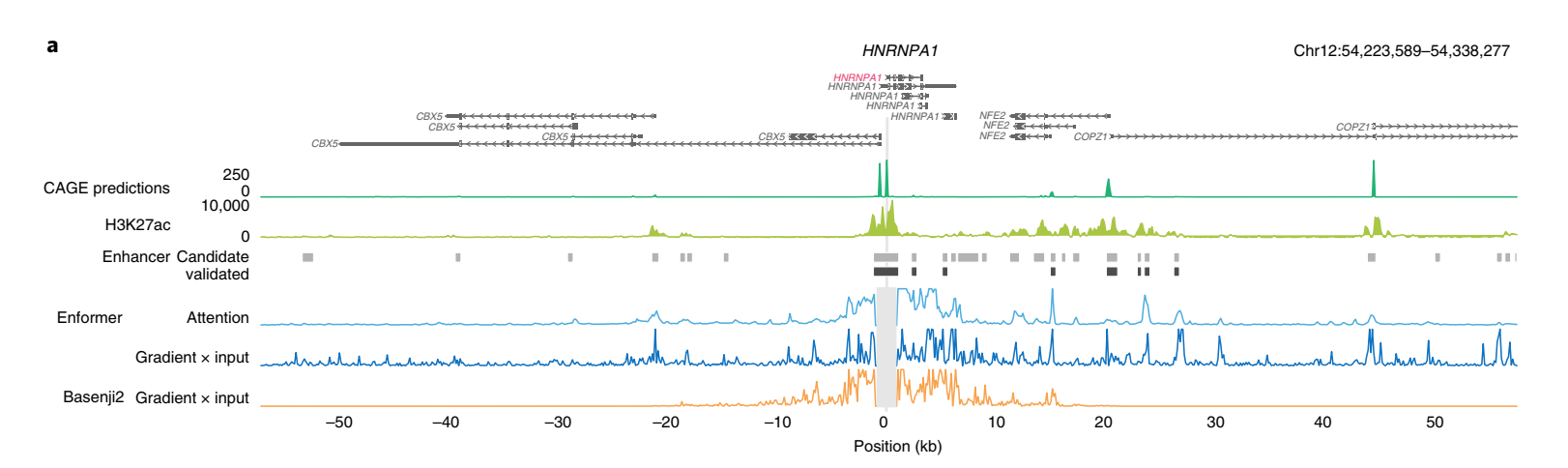
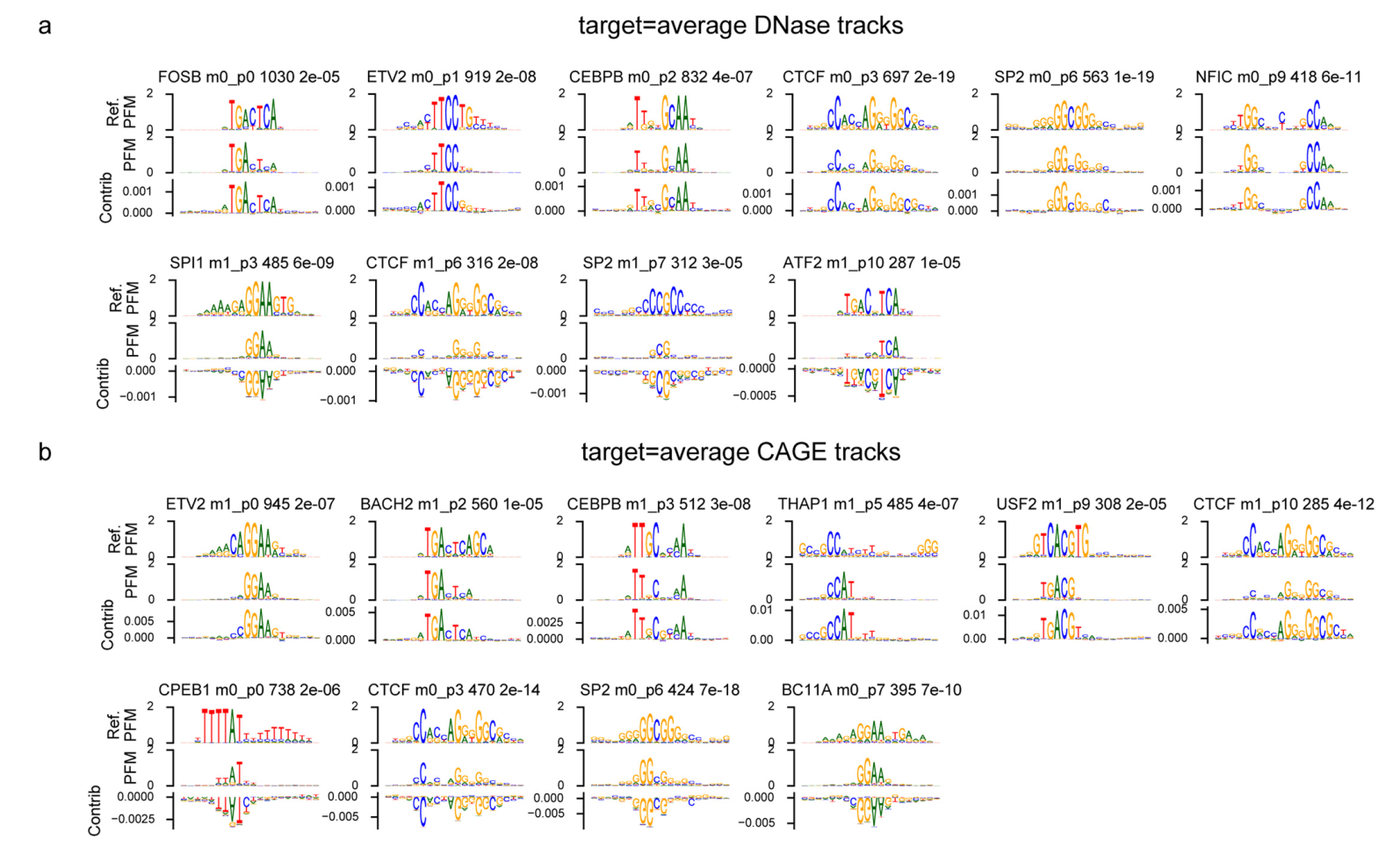
为了更好地理解Enformer在进行预测时使用了哪些序列元件,我们为几个具有经CRISPRi验证的增强子的基因计算了两种不同的基因表达贡献分数——输入梯度(梯度×输入)和注意力权重(方法和补充图1)。贡献分数突出显示了对特定基因表达预测最重要的输入序列。体内突变和梯度×输入是组织或细胞类型特异的,因为它们是相对于特定输出CAGE样本(例如K562)计算的。相比之下,注意力权重是模型的内部特征,在所有组织和细胞类型的预测中共享。
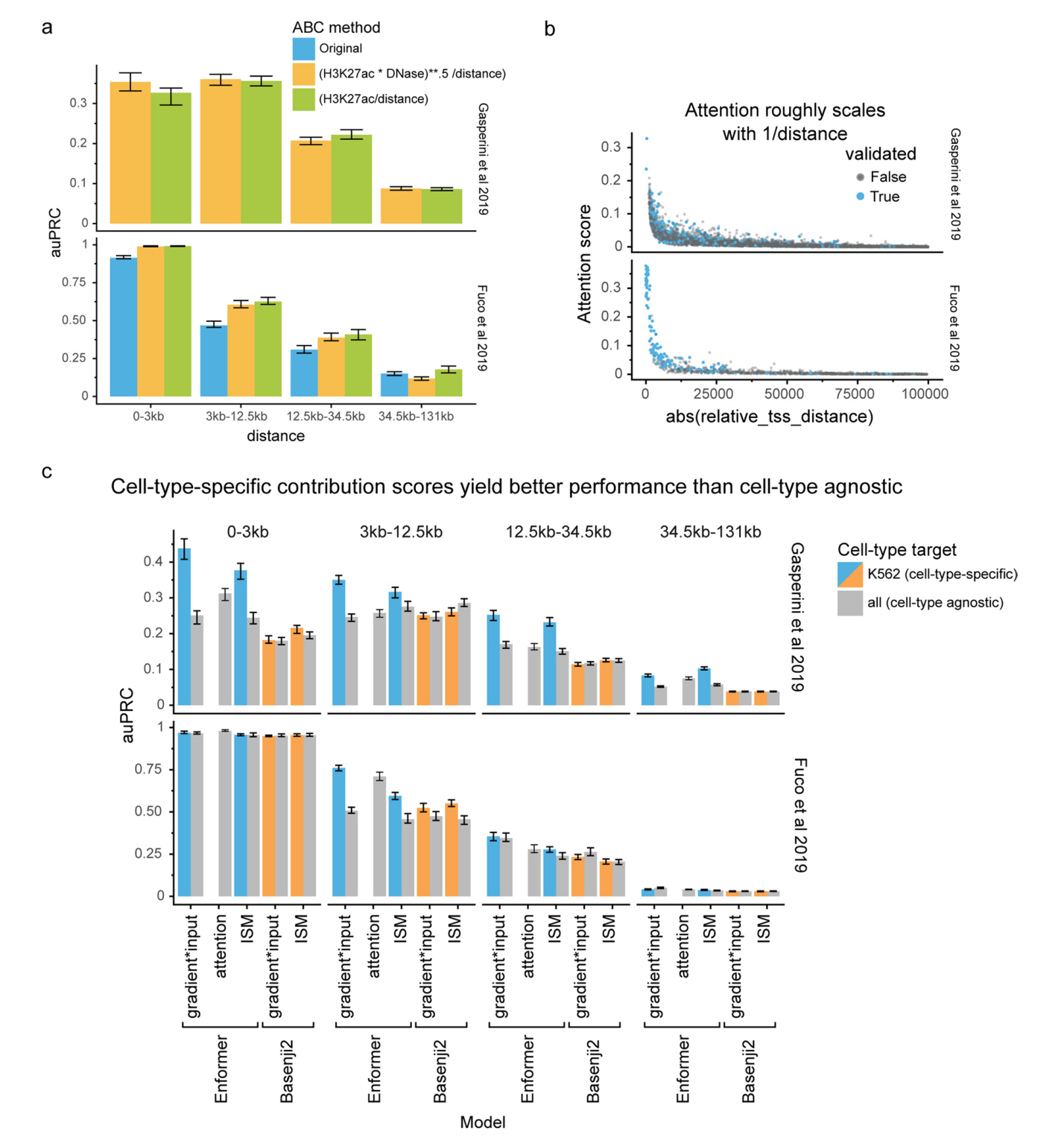
我们检查了几个基因的贡献分数,发现它们与组蛋白H3在K27位乙酰化(H3K27ac)相关,不仅突出了局部启动子区域,还突出了距离超过20 kb的远端增强子(图2a和补充图2、3)。相比之下,由于感受野有限,Basenji2的贡献分数在距转录起始位点20kb以外的序列为零,因此错过了几个增强子。这个例子表明,Enformer在进行预测时确实关注了生物学相关区域,如距离超过20kb的增强子,并且基因表达贡献分数可以用来优先考虑相关的增强子。
H3K27ac:发生在组蛋白 H3 的第 27 位赖氨酸(lysine 27, K27)上的 乙酰化修饰。它是一种表观遗传标记,与基因表达活跃和增强子功能密切相关。
K562:一种来源于人慢性髓性白血病的永生化细胞系,常用于研究血液相关疾病、基因表达调控和分化机制。
将通过生化注释识别的候选增强子链接到靶基因(通俗说:就是找出增强子和它实际调控的基因之间的关系)是一个重要但尚未解决的问题。由于标记的噪声和类别不平衡的组合,计算模型历来产生的准确性较低。为了系统地评估贡献分数在确定特定基因相关增强子方面的能力,我们比较了在K562细胞系上进行的两个大规模CRISPRi研究中测试的所有增强子-基因对的几种贡献分数。在这些实验中,使用CRISPRi抑制了超过10,000个候选增强子的活性,并测量其对基因表达的影响。
Enformer贡献分数在几乎所有相对距离和不同类型的贡献分数上,都比Basenji2贡献分数或随机分数更准确地优先考虑了经验证的增强子-基因对(图2b,Enformer对Basenji2对随机)。Enformer的性能可以与ABC评分(一种最近专门为增强子优先级提出的最先进方法)相媲美,在某些情况下甚至更好。这一点很显著,因为ABC评分依赖于实验数据作为输入,如基于HiC的相互作用频率和H3K27ac(图2b,蓝色对绿色,和扩展数据图7a),而Enformer只使用DNA序列作为输入,并且从未被明确训练来定位增强子。这使得Enformer也可以用于缺乏实验数据的任意序列变异。
ABC评分:用于评估和优先排序基因组中增强子的功能性及其潜在靶基因。它基于增强子和靶基因之间的活性、可及性和三维接触数据,提出的一种定量化的评分体系。
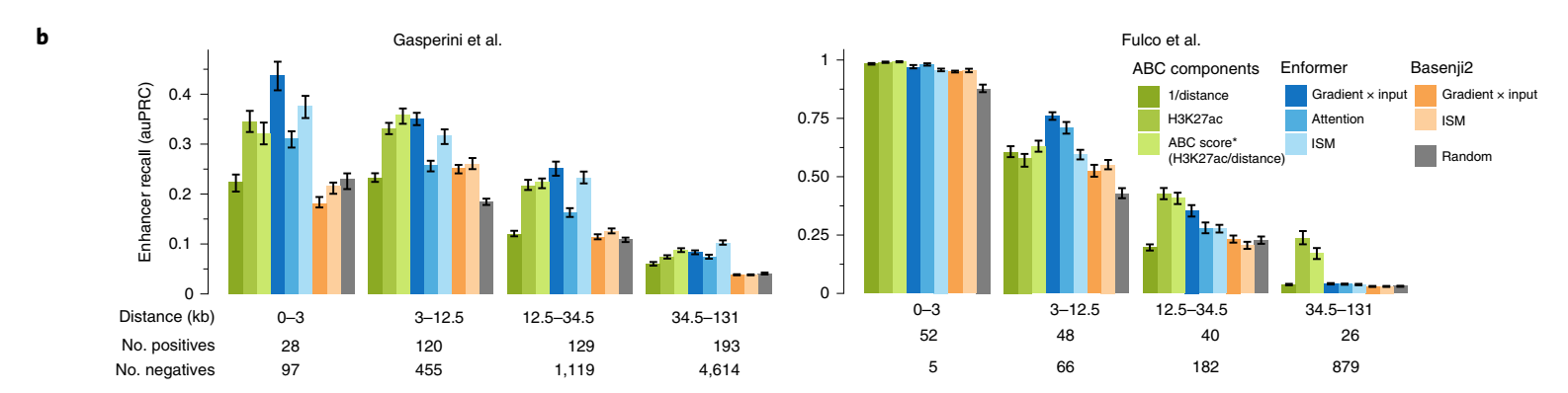
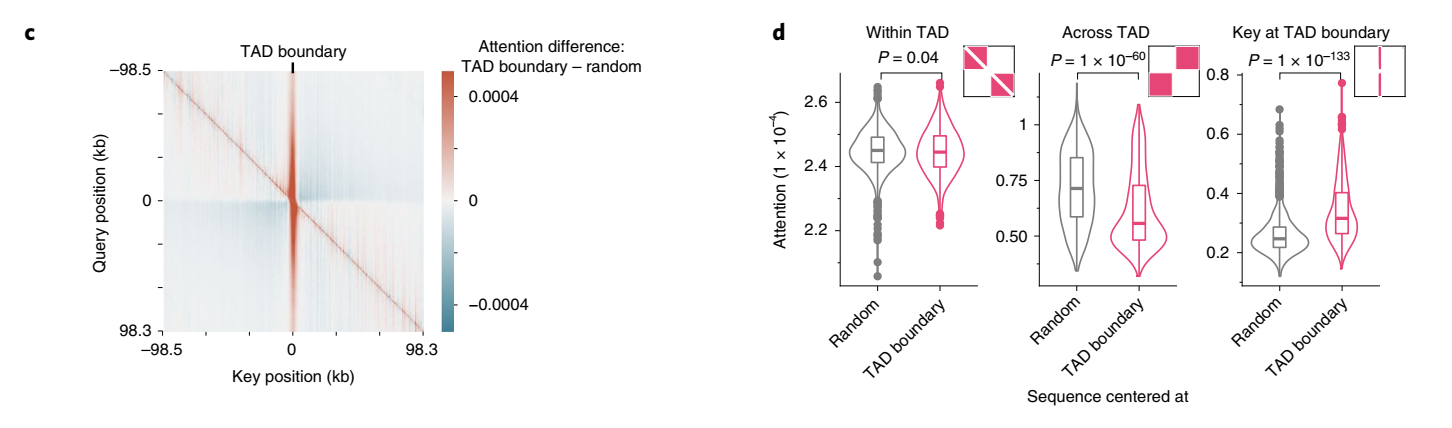
细胞类型特异的贡献分数比细胞类型非特异的贡献分数产生了更高的优先级性能,表明该模型按预期在不同细胞类型中使用不同的增强子序列(扩展数据图7c)。因此,Enformer贡献分数是一种有效的策略,可以在用于模型训练的细胞类型中确定候选增强子的优先级。
接下来,我们探讨了模型是否学习了另一类重要的调控元件:绝缘子元件。这些元件将两个拓扑相关结构域(TAD)分开,并最小化两者之间的增强子-启动子串扰。我们检查了序列中心位于TAD边界处的注意力矩阵(相比于输入梯度,这些矩阵计算更有效,因为输出目标较多),并将其与没有特定对齐的序列的注意力矩阵进行比较。从查询位置的角度来看,Enformer对TAD边界的注意力比对随机位置的注意力更多(垂直红色条纹,图2c),而对边界另一侧区域的注意力更少(对角线外的蓝色区块,图2c),这与生物学上观察到的TAD间相互作用减少的现象一致。在1,500个测试序列中,这两种模式都具有统计学显著性(图2d,"跨TAD"和"TAD边界处的关键位置")。在TAD边界处,模型用于做出DNase和CAGE预测的关键基序之一是CTCF,这种基序被发现与正负贡献分数都有关(扩展数据图8)。总的来说,这些结果表明,该模型不仅学习了组织特异性增强子和启动子的作用,还学习了绝缘子元件及其在抑制基因组区室间信息流动中的作用。
拓扑相关结构域(TAD, Topologically Associating Domains):染色质三维结构中的功能性单位,指的是基因组中形成相对独立的空间区域,区域内的 DNA 片段更倾向于彼此互作,而与区域外的片段的互作较少。这种分区结构在基因表达调控中扮演重要角色。
DNase(脱氧核糖核酸酶):一种酶,能够催化DNA分子的水解反应,将DNA切割成较小的片段。DNase常用于研究染色质的开放状态和基因调控区域,特别是在染色质免疫沉淀(ChIP-Seq)和DNase-seq技术中,帮助识别活跃的基因调控元件。
CTCF(CCCTC-binding factor):一种高度保守的转录因子,广泛存在于真核生物中,尤其在基因调控和染色质结构中扮演重要角色。CTCF主要通过结合DNA上的特定序列(CCCTC)来调控基因的表达。
Enformer改进了eQTL数据的变异效应预测
这项研究的一个核心目标是预测遗传变异对细胞类型特异性基因表达的影响,以帮助解释从全基因组关联研究中发现的数千个与表型相关的非编码关联。计算模型可以处理不同的等位基因并比较预测来评分遗传变异。一个成功的模型应该能够在不需要测量数百到数千个个体基因表达谱的情况下,产生基因表达数量性状位点(eQTL)研究的结果。
eQTL(表达数量性状位点):指在基因组中与基因表达水平变异相关的遗传区域。eQTL分析研究的是特定基因的表达水平如何受到基因组变异(如SNPs)的影响。通过这种分析,可以识别与某些疾病、性状或生物学过程相关的基因调控区域。
因此,我们研究了GTEx项目在数十个人类组织中发现的eQTL来验证模型预测。这种验证的主要挑战在于所研究人群中变异的共现(即连锁不平衡)会将因果eQTL的效应转移到邻近共现变异的测量值上。有向连锁不平衡谱(SLDP)回归是一种技术,它可以在考虑连锁不平衡的情况下,测量有向变异注释(如我们的模型预测)和GWAS汇总统计数据(如GTEx eQTL)之间的全基因组统计一致性(见方法)。在648个CAGE数据集(提供转录本的定量信息)中,有379个(59.4%)数据集中,跨GTEx组织的最大SLDP Z得分(代表最可能的最接近样本匹配)在Enformer预测中相比Basenji2有所增加。相比Basenji2,有228个Enformer最大Z得分增加超过一个标准差,而只有46个减少超过一个标准差。最大Z得分平均从6.3增加到6.9(图3a)。
GTEx(Genotype-Tissue Expression)项目:一个旨在研究基因型与基因表达之间关系的大型国际合作项目。该项目的目标是通过收集来自不同组织的样本,分析基因型(遗传变异)如何影响基因的表达,并探索这些变异如何与健康和疾病相关。GTEx项目收集了来自数百名个体的多种人体组织样本(例如大脑、肝脏、心脏等),对其进行基因组学分析,构建了一个基因型与基因表达的关系数据库。
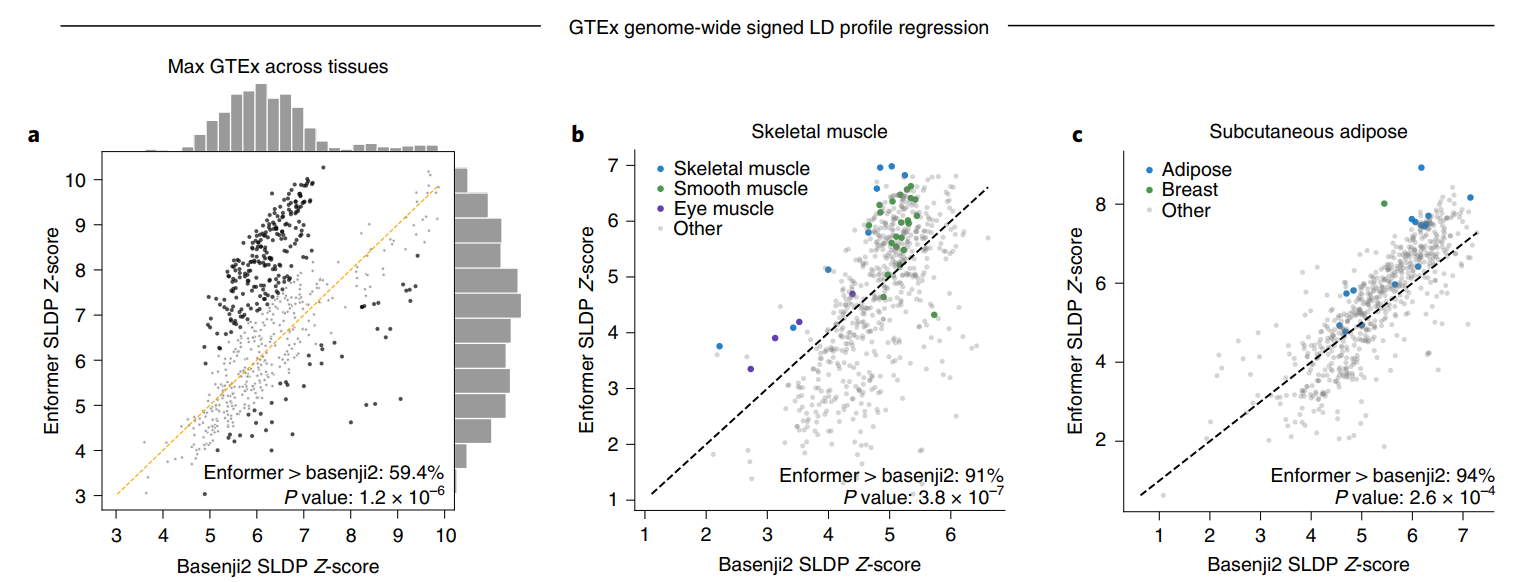
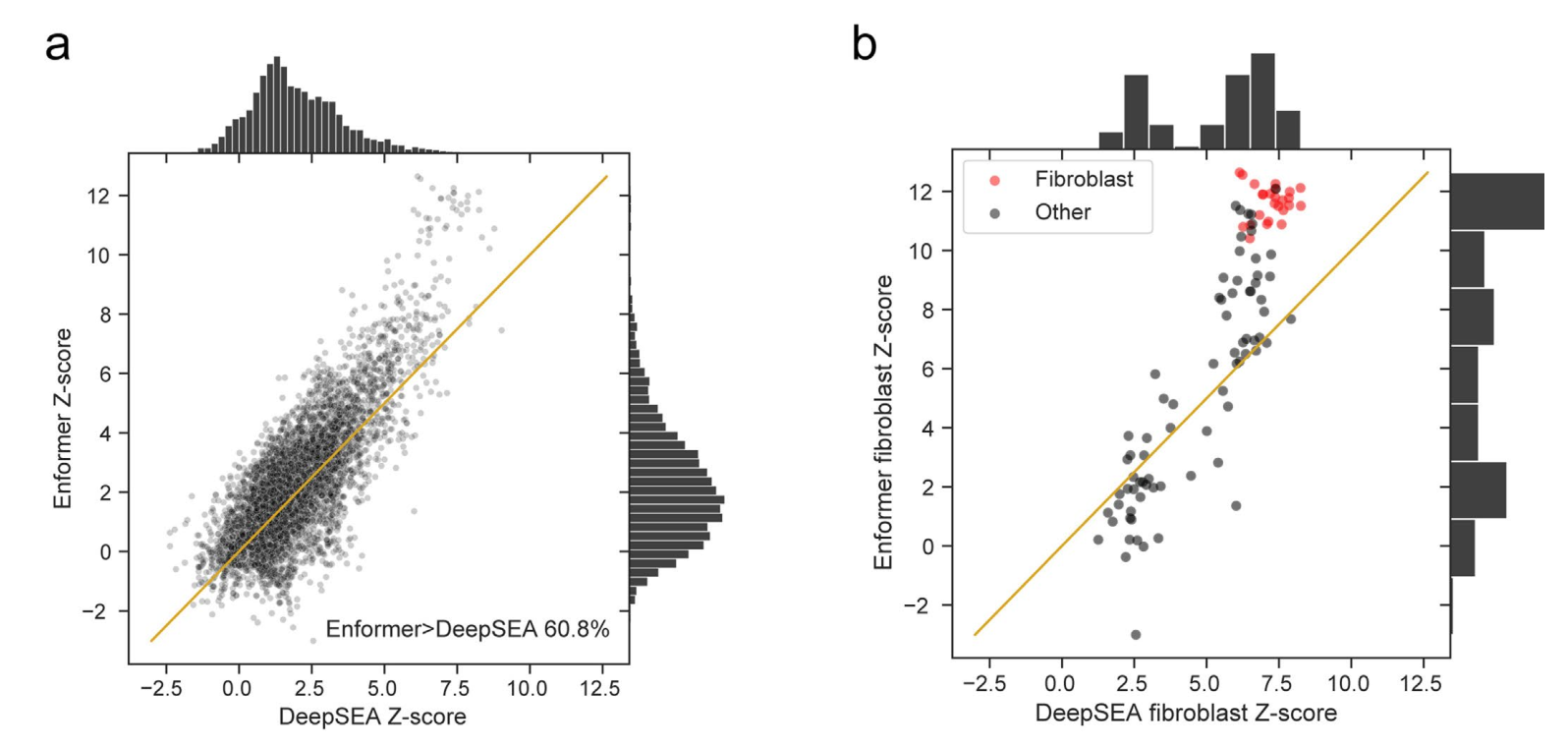
需要注意的是,我们并不期望没有相关GTEx组织匹配的CAGE样本会出现增加的SLDP Z得分。在研究GTEx骨骼肌和皮下脂肪组织的SLDP时,我们观察到生物学相关的CAGE数据集(显示为蓝色)在从Basenji2到Enformer的过程中有所改善(图3b、c)。我们还发现,Enformer对DNase超敏感性的变异效应预测与GTEx的SLDP一致性高于DeepSEA Beluga(在ExPecto中使用)的替代方法(扩展数据图9)。因此,Enformer对非编码变异活性的预测似乎主要在样本具有相似细胞类型组成时得到改善,这与我们观察到的对保留序列的组织和细胞类型特异性改善一致。
虽然连锁不平衡通常导致GTEx eQTL关联只能归因于频繁共现的变异集,但最新的GTEx版本包括了许多具有简单连锁模式的位点中的数千个关联,这些位点已经被精细定位到单个高概率的因果变异。
因果变异:直接引起特定生物学性状或疾病的遗传变异。这些变异不仅与性状或疾病相关联,而且在遗传上具有因果关系,即它们直接影响性状或疾病的发生或发展。
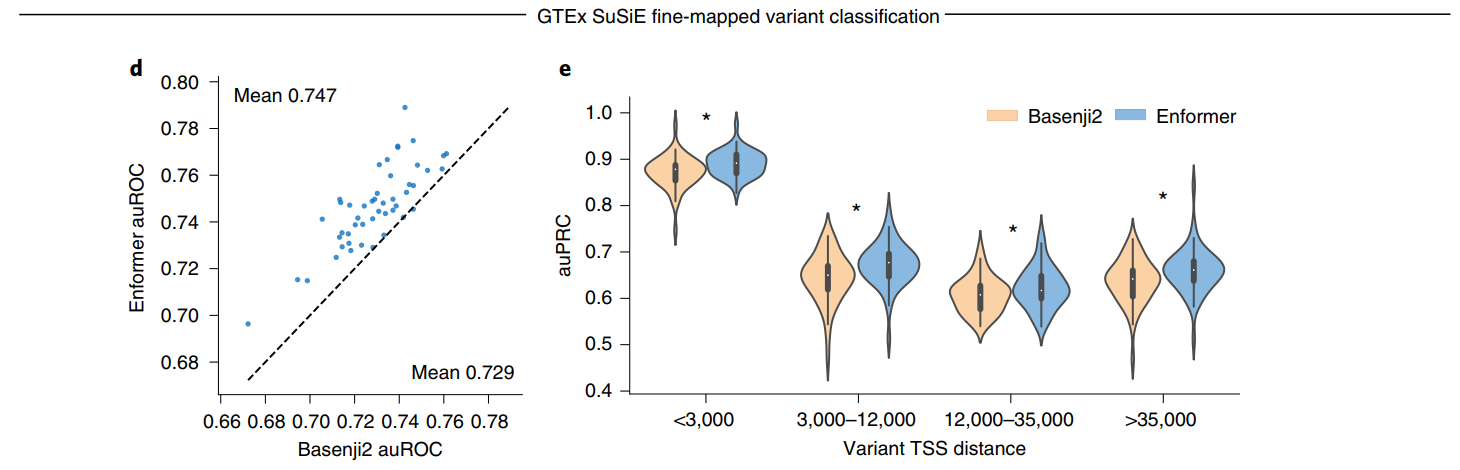
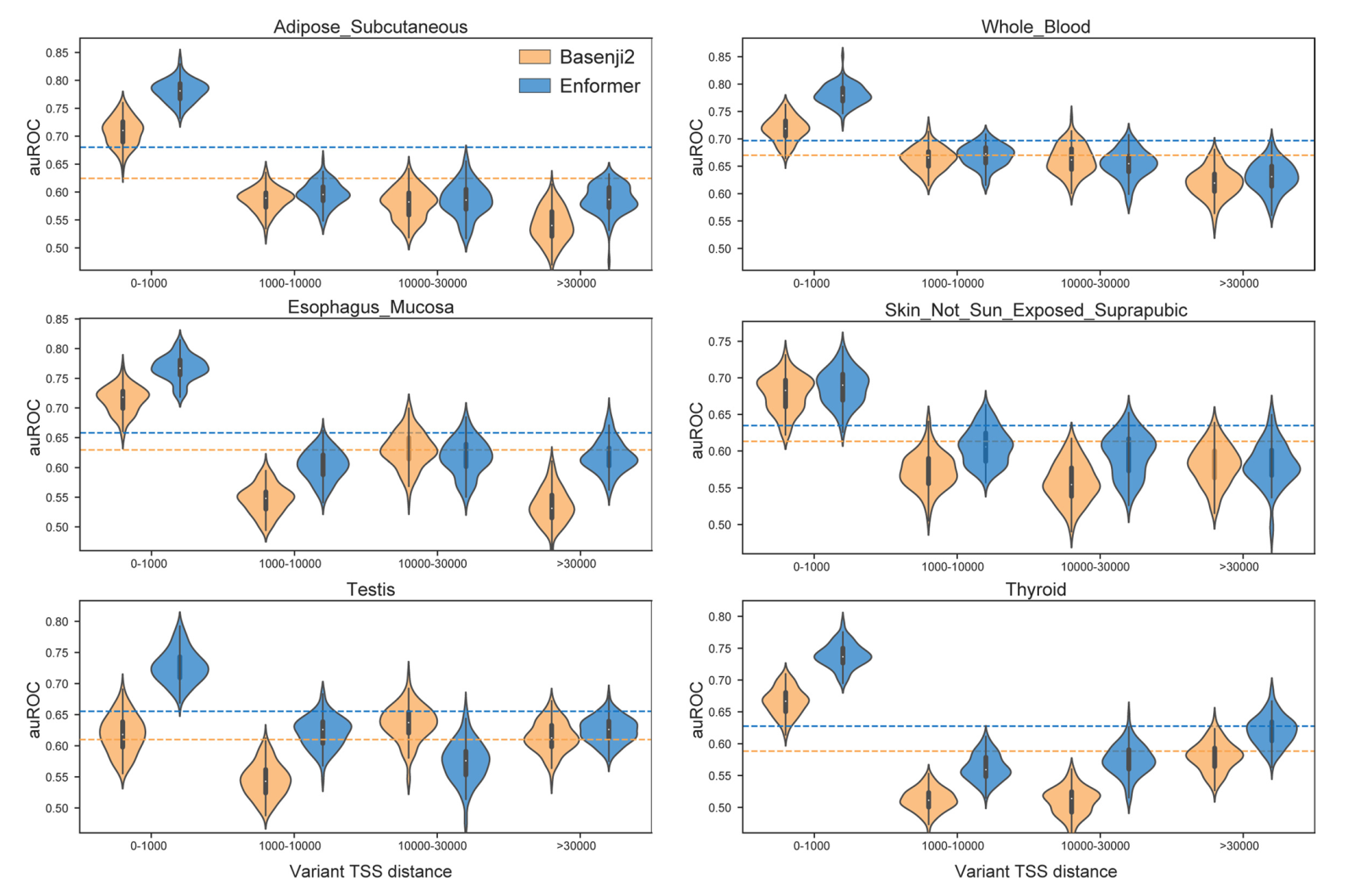
为了评估Enformer预测在识别因果变异方面的效用,我们为每个组织定义了一个分类任务,以区分可能的因果变异(因果概率>0.9,由基于人群的精细定位模型SuSiE确定)和可能的假阳性eQTL(因果概率<0.01),这些变异在可能的情况下与eGene相匹配(见方法)。我们通过计算参考和替代等位基因之间的预测差异向量(即评估参考减去替代等位基因,在序列上求和)来表示每个变异,为所有5,313个人类数据集产生特征。我们训练随机森林分类器。Enformer的预测使得分类器在48个GTEx组织中的47个组织中更加准确(图3d),平均auROC从0.729增加到0.747。这种改进在所有与转录起始位点的距离上都是一致的(图3e),这表明该模型不仅能更好地表示可能重叠长程增强子的变异(通过更大的感受野实现),而且还能更有效地解析启动子和短程增强子。在这些精细定位的eQTL中,Enformer模型在预测表达变化方向方面也比Basenji2更准确(扩展数据图10)。
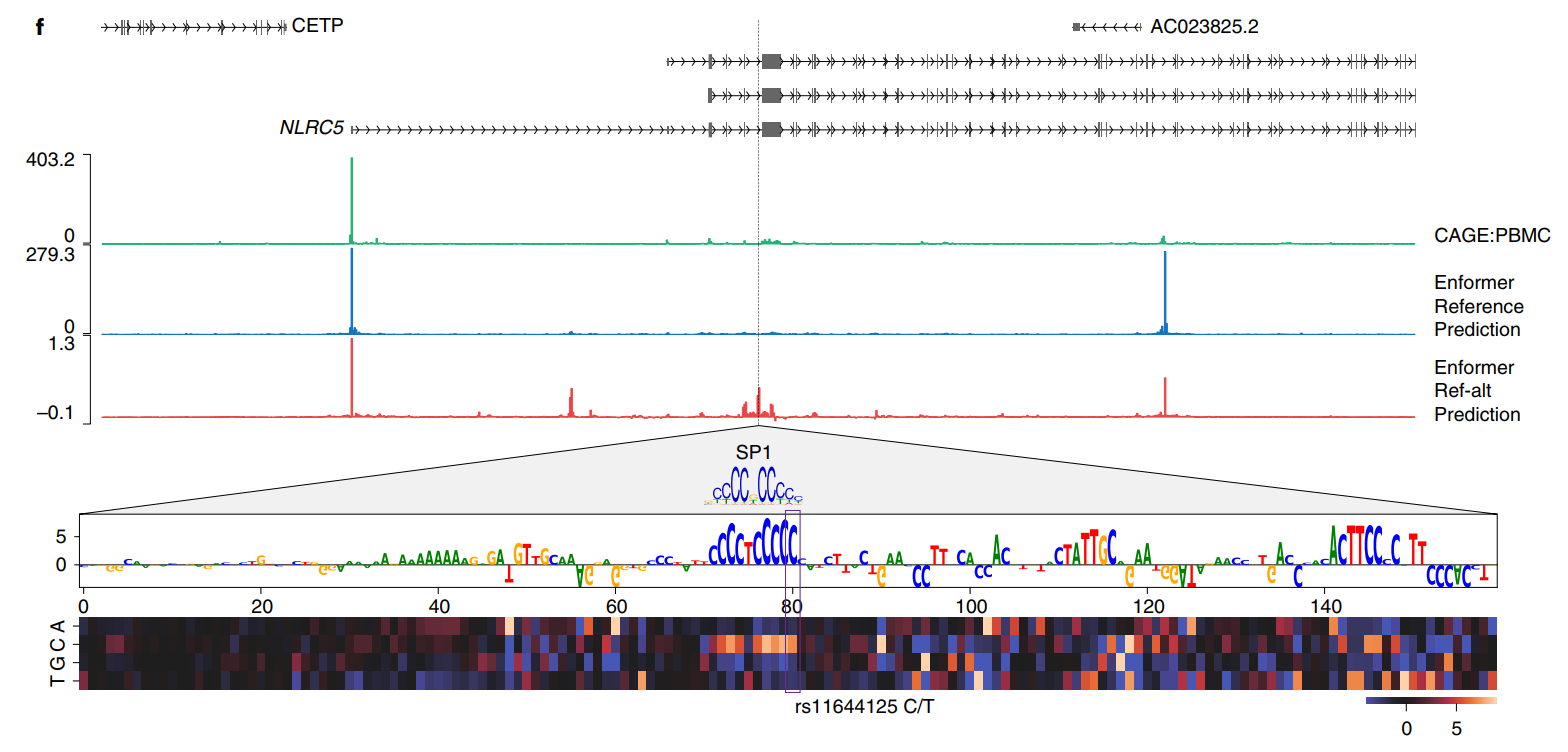
一个Enformer eQTL概率预测相对于Basenji2有所增加的变异示例是rs11644125,它位于NLRC5基因转录起始位点下游约35kb的内含子中,该基因参与病毒免疫和细胞因子反应(图3f)。该变异已经在统计学上被精细定位为可能导致单核细胞和淋巴细胞血细胞计数变化。根据GTEx的数据,相对于主要等位基因C,次要等位基因T降低了全血中NLRC5的基因表达。Enformer正确预测了许多相关CAGE样本(包括外周血单个核细胞)中上游转录起始位点的NLRC5表达降低。使用局部区域的体内突变(见方法),我们观察到变异rs11644125调节了转录因子SP1的已知基序。Enformer预测表明,在造血细胞中SP1结合的扰动改变了NLRC5表达,这是这些性状的一个机制。
Enformer改进了MPRA变异效应预测
MPRA(Massively Parallel Reporter Assays):一种用于高通量评估基因调控元件功能的实验技术。MPRA结合了报告基因系统和高通量测序技术,能够同时评估大量潜在的基因调控元件(如增强子、启动子等)的活动。
最后,我们使用第二个独立的变异效应预测任务评估了Enformer的性能,这个任务使用了一个数据集,其中大规模平行报告基因检测(MPRAs)通过对几个增强子和启动子在各种细胞类型中进行饱和突变,直接测量了遗传变异的功能效应。我们使用了与CAGI5竞赛相同的训练和测试集,使我们能够直接将Enformer的性能与其他团队提交的结果进行基准比较。其他团队采用的方法包括多种不同的方法,从使用deltaSVM策略、CADD框架,到使用结合了保守性信息和来自DeepBind和DeepSEA的深度学习预测的特征的回归模型(第3组、第5组和第7组)。对于每个变异,我们通过计算参考和替代等位基因之间的预测差异来评估其效应,获得了5,313个特征。接下来,我们比较了两种方法:
- 我们使用这些特征在每个基因提供的训练集上训练
lasso回归模型; - 我们预先选择了与细胞类型匹配和细胞类型非特异的
CAGE和DNase变化预测相对应的特征子集,并生成了特征的统计摘要(即无需额外训练)。
在每个基因的测试集上评估这两种方法揭示,使用Enformer预测作为特征的lasso回归在所有位点中平均具有最佳相关性,超过了七种替代方法(图4a)。
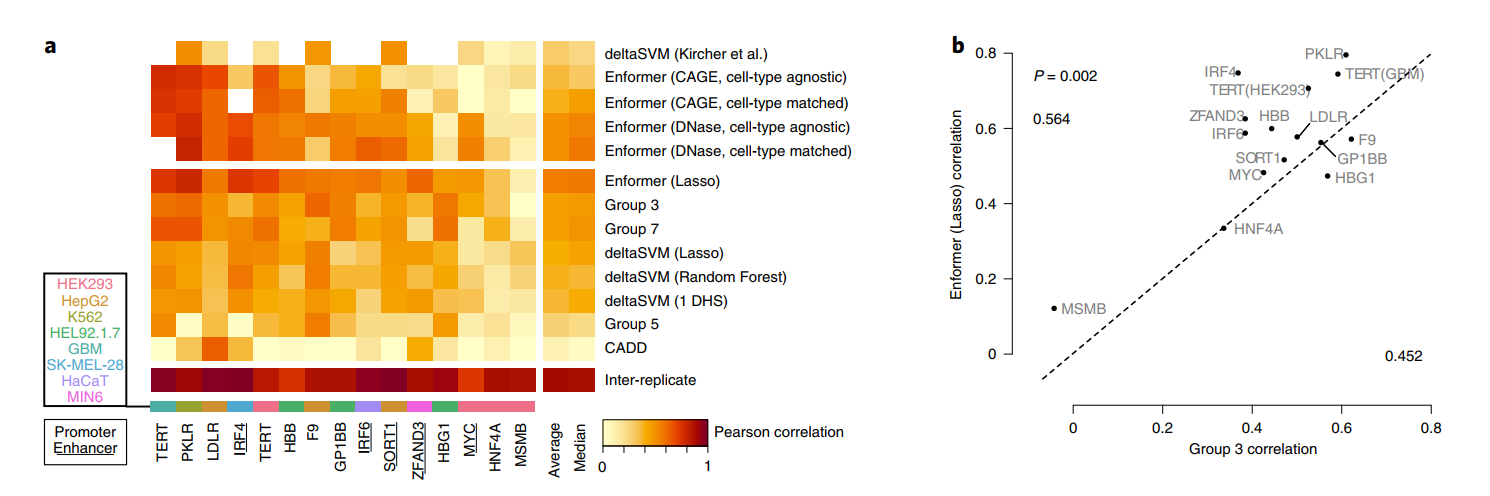
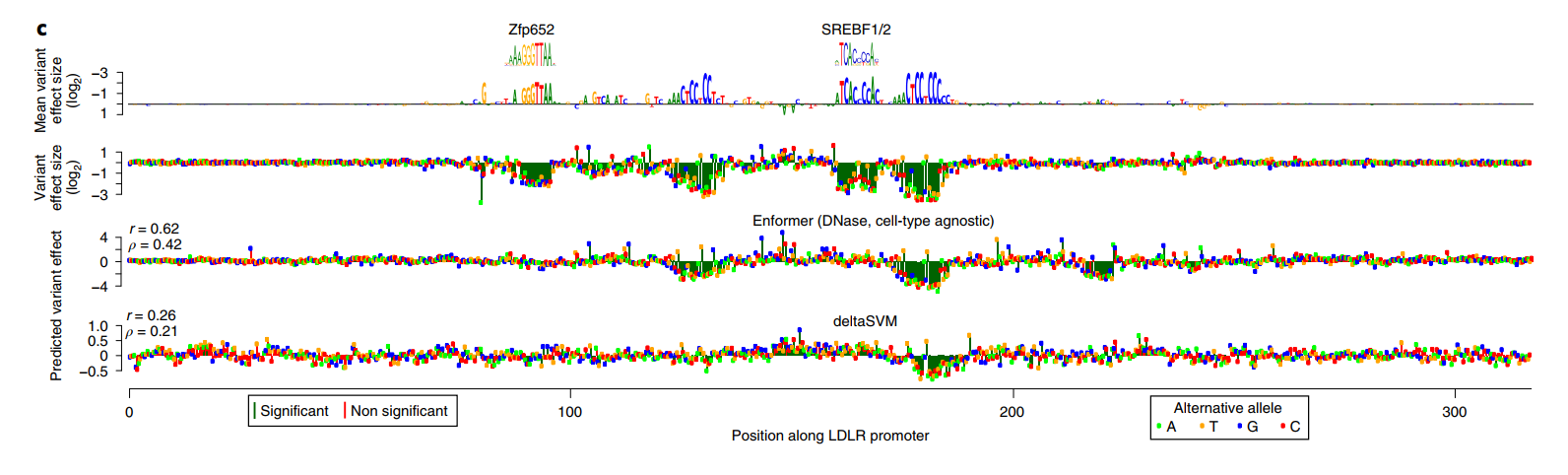
此外,无需训练的Enformer预测直接用作评分时,其性能与经过lasso训练的模型相当,同时也优于其他提交的方法。这包括基于序列的预测器deltaSVM,该预测器是在来自匹配细胞类型的独立DNase和组蛋白H3在K4位单甲基化(H3K4me1)数据上训练的。经过lasso训练的Enformer超过了CAGI5获胜团队第3组的性能(P=0.002,配对、单侧Mann-Whitney U检验,图4b)。对不需要额外训练的预测进行可视化表明,Enformer忠实地捕捉到了LDLR位点中四个转录因子结合位点中的两个的效应(图4c)。
Enformer突出显示了一个额外的结合位点,虽然它的效应大小较小,但仍显示出显著差异。相比之下,deltaSVM只成功预测了一个结合位点但错过了其他三个,总体上相对于Enformer表现出50%降低的Pearson和Spearman相关性。对于这个位点,细胞类型匹配的预测反映了细胞类型非特异的预测,表明检测到的结合位点可能对应于大多数细胞类型中存在的一般转录因子。
讨论
监管基因组学中一个长期存在的问题是如何仅从DNA序列预测基因表达。通过一种新颖的transformer架构,我们通过大大扩展感受野并增加远距离元件之间的信息流动,取得了重要进展。通过这种方式,该模型可以更好地捕捉生物学现象,例如增强子调控启动子,即使两者之间存在较长的DNA序列距离。这导致组织和细胞类型特异性基因表达预测相关性显著提高,从0.81提高到0.85,向实验水平精确度0.94(从重复实验估计)迈进了三分之一的距离。
这种预测准确性的提高转化为两个具有生物学相关性的关键问题的改进模型:增强子-启动子预测和非编码变异效应预测。我们观察到模型在做出基因表达预测时关注增强子并考虑绝缘子,这表明它已经学会了典型的远距离调控模式。使用Enformer模型,我们可以比以前的方法更准确地预测自然变异或CRISPR干扰的增强子是否会导致显著的表达变化。通过仅依赖DNA序列作为输入,Enformer相比替代的变异效应预测方法具有几个优势:
- 与大多数方法不同,它能够对激活或抑制性突变进行有向预测;
- 由于不明确依赖核苷酸保守性统计数据(大多数工具都依赖),其预测不限于保守的增强子,而保守的增强子仅占所有增强子的一小部分;
- 它可以对任意序列进行预测,这使得可以合成设计具有细胞类型特异性的增强子。
总的来说,这些进展和优势为研究与疾病相关的遗传变异不断扩大的目录以及发育和进化中的增强子生物学开辟了令人兴奋的途径。
有几条路径可以进一步提高模型准确性,这些路径看起来很有希望。机器学习的成功取决于训练数据,因此提高目标轨迹的分辨率和质量,以及收集来自其他生物体的数据,可能会提升性能。最近的工作表明,高度结构化的3D DNA接触(对长程基因调控有重大影响)可以从基础DNA序列预测。巧妙地将这些模型与我们的模型结合可能会改善Enformer对绝缘子和远距离调控的建模。
当前方法的一个限制是我们只能为训练数据中的细胞类型和检测方法进行建模和预测,而无法推广到新的细胞类型或检测方法。并行研究已经开始通过细胞类型和检测方法的表征学习来解决这个缺点,并可能在未来使用Enformer架构。该模型对遗传变异的敏感性可以通过在越来越多的功能基因组数据集(如来自CRISPR干扰和大规模平行报告基因检测的数据集)上进行训练来进一步改善。目前,这些数据集的小规模限制了它们仅用于模型评估。最后,我们预计transformer模型最近在计算效率方面的改进以及更好的硬件将使我们能够进一步扩展模型规模。
未来,Enformer可以被系统地应用于精细定位现有的GWAS研究,优先考虑罕见疾病中观察到的罕见或从头突变,并跨物种推断调控活性以研究顺式调控进化。为了促进这些下游应用,我们已经公开发布了预训练的Enformer模型,并提供了演示其使用方法的代码示例。此外,我们已经预先计算了1000个基因组数据集中所有常见变异的效应预测,并公开发布。我们希望我们的模型将促进对基因调控架构的更好理解,并推动改进用于基因起源疾病诊断工具的开发。
方法
模型架构
Enformer架构由三个部分组成:
- 带有池化的7个卷积块;
- 11个
transformer块; - 1个裁剪层,后跟最终的逐点卷积,分支成2个生物体特异的网络头(扩展数据图1)。
Enformer接受长度为196,608 bp的独热编码DNA序列作为输入(A = [1,0,0,0],C = [0,1,0,0],G = [0,0,1,0],T = [0,0,0,1],N = [0,0,0,0]),预测人类基因组的5,313个基因组轨迹和小鼠基因组的1,643个轨迹,每个长度为896,对应于聚合成128 bp区块的114,688 bp。带有池化的卷积块首先将空间维度从196,608 bp减少到1,536,使得每个序列位置向量代表128bp(尽管卷积也观察相邻池化区域中的核苷酸)。
transformer块然后捕获序列中的长程相互作用。裁剪层在每侧修剪320个位置,以避免在远端计算损失,因为这些区域处于不利地位,它们只能观察一侧(朝向序列中心)而不能观察另一侧(序列边界以外的区域)的调控元件。
最后,两个输出头预测生物体特异的轨迹。Enformer架构与最先进的模型Basenji2类似。然而,以下改变帮助我们改进并超越了其性能:Enformer使用transformer块而不是扩张卷积,使用注意力池化而不是最大池化,使用两倍的通道数,以及1.5倍长的输入序列(197kb而不是131kb)。
详细的模型架构,包括选定的超参数,如扩展数据图1所示。
注意力池化通过以下方式总结输入序列 在个位置上的连续块,对于 个通道中的每一个返回输出值 :
其中 索引池化窗口中的序列位置,由指数化的点积 加权,是学习的权重矩阵。我们对原始输入序列的连续块应用注意力池化,使用窗口大小 和步长为2。我们将 初始化为,其中是单位矩阵,以优先考虑较大的值,使该操作类似于最大池化。这种初始化比随机初始化或用零初始化(表示平均池化)的性能略好。
我们使用多头注意力(MHA)层在序列中共享信息并模拟长程相互作用,例如启动子和增强子之间的相互作用。每个头都有一组独立的权重 、和,它们将输入序列 转换为查询、键和值。查询表示每个位置的当前信息,键表示每个位置将寻找的信息。它们的点积加上相对位置编码 形成注意力矩阵,计算如下:
其中条目 表示位置i处的查询对位置j处的键的权重。值表示每个位置将传播给关注它的位置的信息。每个单独的注意力头计算其输出为跨所有输入位置的加权和:。这允许每个查询位置使用整个序列的信息。多个头使用独立的参数进行计算,我们将每个头的输出连接起来形成最终的层输出,然后是一个线性层来组合它们。我们的层使用8个头,值大小为192,键/查询大小为64。
在自然语言处理中的MHA应用通常直接在输入序列上操作,将其标记为单词并嵌入到更丰富的嵌入空间中。Enformer模型中位于MHA之前的卷积塔用于执行类似的操作,即嵌入核苷酸片段,并为相邻核苷酸在基序中一起工作提供了令人信服的归纳偏置。我们选择在128bp分辨率下计算,因为它大致代表了一个包含几个基序的调控元件的研究长度,并且是聚合实验数据进行预测的适当区块大小。更精细的分辨率在数据支持时可能有潜在的好处,但会延长进入二次复杂度MHA的序列长度,使模型工程在当前可用的硬件上变得难以处理。
为了注入位置信息,我们按照Transformer-XL论文中的公式,将相对位置编码 添加到 注意力项中。具体来说,我们使用:
其中 是不同相对基函数 的线性函数, 和 是用于评估对特定键()或相对距离()偏好的位置无关嵌入。我们使用三种不同的基函数类用于 ,如扩展数据图5b所示。
-
指数函数:
其中 在对数空间中在3和序列长度之间线性放置。
-
中心掩码函数:
-
伽马函数:
其中 是伽马概率分布函数。 在(序列长度/特征数量)到序列长度之间线性放置,=序列长度/(2 × 特征数量)。
对于每个基函数,我们使用对称 和非对称 版本来引入方向性。我们使用与MHA的值大小相同数量的相对位置基函数(192)。这192个基函数在基函数类和对称与非对称版本之间平均分配。对于3个基函数类,每个基函数类提供64个位置特征(32个对称和32个非对称)。
在MHA中,位置编码特征和最终注意力矩阵的丢弃率分别为0.01和0.05。所有其他丢弃率都在扩展数据图1a中注明。
模型训练和评估
模型在与Basenji2相同的目标、基因组区间和泊松负对数似然损失函数上进行训练、评估和测试。简而言之,跨物种训练/验证/测试集是通过以下程序构建的,以将同源序列划分到相同的集合中。首先,我们将人类和小鼠基因组分成1Mb区域。我们构建了一个二分图,其中顶点表示这些区域。接下来,如果两个区域在从UCSC Genome Browser下载的hg38-mm10同源网格格式对齐中具有超过100kb的对齐序列,我们就在它们之间放置边。最后,我们将二分图中的连通分量随机划分为训练集、验证集和测试集。
数据集包含人类基因组的34021个训练序列、2213个验证序列和1937个测试序列,以及小鼠基因组的29295个训练序列、2209个验证序列和2017个测试序列。对于人类基因组,每个样本包含2131个转录因子(TF)染色质免疫沉淀测序(ChIP-seq)、1860个组蛋白修饰ChIP-seq、684个DNase-seq或ATAC-seq和638个CAGE轨迹(总计5313个,补充表2)。对于小鼠基因组,每个样本包含308个TF ChIP-seq、750个组蛋白修饰ChIP-seq、228个DNase-seq或ATAC-seq和357个CAGE轨迹(总计1643个,补充表3)。我们修改了Basenji2数据集,使用hg38参考基因组将输入序列从原来的131072bp扩展到196608bp。
为了在人类和小鼠基因组上同时训练模型,我们在包含人类基因组数据的批次和包含小鼠基因组数据的批次之间交替。主要的Enformer模型具有1536个通道,使用Sonnet v2、TensorFlow(v2.4.0)实现,并在64个TPU v3核心上进行训练,批量大小为64(每核心1个),训练150000步(约3天),在每一步中使用全归约梯度聚合。批量归一化统计数据也使用0.9动量在多个副本之间聚合。我们使用来自Sonnet v2的Adam优化器,学习率为0.0005,其他超参数使用默认设置:。
通过网格搜索在验证集上获得最高性能来发现最佳学习率。我们在训练的前5000步中将学习率从0线性增加到目标值。我们将梯度裁剪到最大全局范数0.2。在训练期间,我们使用与Basenji2相同的数据增强,通过随机移动输入序列最多3bp并反向互补输入序列的同时反转目标。最后,我们使用较低的学习率0.0001在人类数据上对Enformer模型进行了30000步的微调。
我们使用预训练的Basenji2模型进行所有主要的模型比较,并重新训练了一个等效模型用于消融实验和超参数扫描(如扩展数据图5所示)。在这些比较分析中,我们使用了768个通道(原始Enformer模型的1/2,在MHA中使用值大小96)、131kb输入序列,以及批量大小32在32个TPU v3核心上训练。我们没有在人类数据上对这些模型进行微调。
对于使用扩张卷积而不是transformer块的模型,我们使用了更高的学习率0.02,且不进行学习率预热。与Enformer一样,最佳学习率是通过网格搜索在验证集上获得最高性能来确定的。所有模型都训练了500000步,每1000步在验证集上计算一次CAGE TSS基因表达的Spearman相关性(跨基因平均,跨实验平均),只保存具有最高相关性的模型。
我们使用验证集进行超参数选择,使用测试集进行Basenji2比较。我们考虑了两个评估指标:
-
对于每个输出轨迹,在验证/测试集中所有
128bp分箱基因组位置上计算的Pearson相关性; -
验证/测试集中所有蛋白质编码基因的
CAGE基因表达值( 转换并在每个实验的基因间标准化)的Pearson相关性,可以是每个CAGE实验跨基因计算(主要指标),也可以是跨CAGE实验为每个基因计算(如图1b所示)。
观察到的和预测的基因表达值是通过在基因的所有唯一TSS位置上对观察到/预测的CAGE读数进行求和获得的。对于每个TSS位置,我们使用与TSS重叠的128bp分箱以及两个相邻分箱(总共3个分箱)。
在模型评估期间,我们使用测试时增强:我们平均了8个随机增强序列的预测,增强方式与训练期间相同(≤3bp移位和反向互补)。我们仅在生成图1时在测试集上评估了模型性能一次,在模型开发期间未使用测试集。
为了选择一个具有代表性的例子,我们将Enformer和Basenji2在"跨CAGE实验"指标上表现差异最大的前10个转录本可视化,该指标用于测量33%组织特异性最强的基因的组织特异性。我们选择了列表中的第六个转录本(ENST00000524922),因为它清晰地显示了所有三类基因组轨迹(DNA可及性、组蛋白修饰和基因表达)的差异。
增强子优先级排序
我们从两项研究中获得了一组增强子-基因对,这些研究使用CRISPRi测定方法扰动目标增强子并测量K562细胞中基因表达变化:Gasperini等人使用scRNA-seq测量表达变化,以及Fulco等人使用Flow-FISH。
我们使用UCSC liftOver网络工具将增强子和基因坐标从hg19转换到hg38。每个增强子-基因对都包含一个标签,表明在CRISPRi处理后是否诱导了显著的表达变化。我们将所有增强子集合表示为"候选"增强子,将那些显示表达变化的增强子表示为"验证"增强子。
我们使用精确度-召回曲线下面积(auPRC)评估不同方法对表现出显著表达变化的增强子-基因对进行分类或优先级排序的能力。
要使用基于序列的模型对增强子-基因对进行优先级排序,我们计算了三种不同的得分:梯度×输入、注意力和体外突变(ISM)。对于每个增强子-基因对,我们通过取Enformer在K562中预测的最高CAGE值来确定基因的主要TSS。我们提取以主要TSS为中心的DNA序列,并计算以下不同的增强子-基因得分:
-
梯度×输入:我们计算CAGE目标(使用K562特异性CAGE目标或所有CAGE目标,扩展数据图7c)在TSS处相对于输入参考序列核苷酸的梯度的绝对值。注意,由于我们的输入序列是独热编码的,取非零通道(参考核苷酸)的输入梯度,等同于计算梯度×输入归因。我们注意到"TSS处的CAGE"始终意味着从三个相邻分箱中求和绝对梯度值,这在基因聚焦的模型评估中也是如此。三个分箱包括与TSS重叠的分箱和两侧各一个相邻分箱。增强子-基因得分是通过在以增强子为中心的2kb窗口中求和绝对梯度×输入得分获得的。 -
注意力:我们首先在所有头部和层之间平均transformer注意力矩阵。我们提取与位于TSS的查询索引对应的行,这样键就对应于不同的空间位置,注意力值指定模型在为TSS做预测时对这些位置的关注程度。我们仅为Enformer计算此贡献得分。增强子-基因得分是通过在以增强子为中心的2kb窗口中求和注意力得分获得的。 -
ISM:体外突变增强子-基因得分是通过比较参考序列与修改序列(其中2kb增强子序列被随机序列替换)在TSS处的K562 CAGE预测来计算的:|f(modified) – f(reference)|。
为了复现Fulco等人引入的ABC得分,我们从ENCODE获取了K562中的H3K27ac ChIP-seq数据(文件访问号ENCFF779QTH)和DNase数据(文件访问号ENCFF413AHU和ENCFF936BDN)的BigWig文件。我们对重复样本的归一化读数进行求和。对于每个轨迹和增强子,我们在以增强子为中心的固定2kb窗口中求和信号。与原始ABC得分使用的约500 bp可变窗口大小相比,这种固定且更宽的窗口产生了更好的性能(扩展数据图4a)。
GTEx SLDP
我们通过使用参考等位基因和替代等位基因对模型进行正向传递,计算它们的差异,并在序列上求和以获得每个训练数据集的带符号得分,来预测遗传变异对各种注释的影响。我们对使用正向和反向互补序列以及向左和向右的小序列移动计算的得分进行平均。我们计算了所有1000 Genomes SNP的得分。
我们使用SLDP来估计这些得分与GTEx v7a的48个组织的汇总统计数据之间的功能相关性,同时考虑了人群连锁不平衡结构(补充信息)。
精细定位的GTEx分类
为了在不需要考虑LD的情况下研究特定的eQTL,我们使用SuSiE方法研究了GTEx v8的统计精细作图。我们关注在可信因果集合中后验包含概率(PIP)大于0.9的变异,从黑质的最少166个变异到胫骨神经的2740个变异不等。我们安排了一个分类任务,以区分这些阳性因果变异和匹配的阴性变异集。
在可用时,我们为每个因果变异从同一基因中测试的PIP<0.01但|Z-score| > 4的集合中选择匹配的阴性变异。当在同一基因中不可用时,我们从全基因组范围内PIP<0.01且|Z-score| > 6的集合中选择。
为了确定不同变异注释的信息量,我们为每个组织训练了单独的随机森林分类器,使用八折交叉验证来区分因果和非因果变异。我们选择了scikit-learn 0.22实现的默认超参数,因为修改它们后准确性提升可以忽略不计。然而,由于来自训练数据集的特征数量庞大,将每个决策树分裂时考虑的最大特征数设置为特征总数的log2大大提高了计算效率。我们进行了100次随机交叉验证洗牌和随机森林拟合迭代,以描绘模型准确性的低方差估计。我们通过比较8×100个不同测试集auROC来对两个不同模型特征集进行统计检验。
对于带符号的GTEx分析,我们基于模型预测区分增加和降低基因表达的因果变异的能力来进行基准测试。在这个分析中,我们去除了对不同顺式基因的基因表达影响方向相反的变异。我们手动将FANTOM5 CAGE样本描述与GTEx组织匹配。我们跳过了具有超过三个可能匹配的情况。在有两个或三个可能匹配的情况下,我们选择了Basenji2和Enformer预测之间平均一致性最好的CAGE样本。我们通过根据该样本的带符号预测对因果变异进行排序来计算auROC统计量。
基于饱和突变数据对变异效应预测进行基准测试
我们从CAGI5竞赛获得了训练集和测试集以及各参赛者的预测准确性(M.Kircher,个人通信,https://genomeinterpretation.org/content/expression-variants)。对于每个变异和位点,我们将其效应评估为参考等位基因和替代等位基因之间的预测差异,在代表512bp的四个相邻分箱中求和,基于人类数据集生成5313个特征。
在计算这个差异之前,所有CAGE特征都在添加伪计数1后进行对数转换。对于每个等位基因,我们对正向和反向互补序列的预测进行平均。我们使用训练集特征计算的缩放因子对测试集的特征进行缩放,使训练特征具有均值0和标准差1。遵循我们之前的工作,我们然后使用这些特征和相应的训练集为每个位点训练一个lasso回归模型。正则化强度由单个λ参数控制,使用R语言中glmnet库的cv.glmnet函数通过对每个训练集进行十折交叉验证来优化。
对于我们的免训练比较,我们选择了对应于细胞类型匹配和细胞类型无关的CAGE和DNase变化预测的特征子集。对于细胞类型无关模型,我们使用了所有638个CAGE或674个DNase特征的子集(补充表2)。对于细胞类型匹配模型,我们还要求CAGE/DNase特征包含以下子字符串:
- 对于
F9、LDLR和SORT1使用"HepG2"; - 对于
GP1BB、HBB、HBG1和PKLR使用"K562"; - 对于
HNF4A、MSMB、TERT(在HEK293T细胞中进行)和MYCrs6983267使用"HEK293";
对于几个位点,并不存在完全匹配的DNase或CAGE样本。因此,我们根据以下子字符串选择最接近的匹配特征:
- 对于
ZFAND3使用"胰腺"; - 对于
TERT(在GBM细胞中进行)使用"胶质母细胞瘤"; - 对于
IRF6使用"角质形成细胞"; - 对于
IRF4使用"SK-MEL";
对于每个位点,我们提取匹配上述子字符串的特征,并使用所示特征的第一主成分(PC)作为我们的汇总统计量,如果PC与特征均值负相关则反转其符号。
数据可用性
基因注释从https://www.gencodegenes.org/(v32)获取。Basenji2训练、验证和测试数据从https://console.cloud.google.com/storage/browser/basenji_barnyard/data获取。
Fulco等人2019的处理后的CRISPRi数据从补充材料获取,Gasperini等人2019的数据从GEO登录号GSE120861获取。用于图2分析的K562中的H3K27ac ChIP-seq数据从https://www.encodeproject.org/获取,文件登录号为ENCFF779QTH,DNase数据的文件登录号为ENCFF413AHU和ENCFF936BDN。由Fudenberg等人2020处理的TAD边界从https://console.cloud.google.com/storage/browser/basenji_hic/insulation获取。精细定位的eQTL可从Wang等人2021的补充材料获取,阴性集从https://console.cloud.google.com/storage/browser/dm-enformer/data/gtex_fine获取。我们从CAGI5竞赛获得了训练集和测试集以及各参赛者的预测准确性(M.Kircher个人通信,https://genomeinterpretation.org/content/expression-variants)。为了与ExPecto进行比较,我们使用了来自https://github.com/FunctionLab/ExPecto/tree/master/resources提供的数据。
代码可用性
我们的核心算法的所有组件,包括完整的模型架构和用于训练和评估模型的示例代码,均可在以下URL下以开源Apache 2.0许可证获取:https://github.com/deepmind/deepmind-research/tree/master/enformer。该代码也已存档在Zenodo:https://doi.org/10.5281/zenodo.5098375。此外,模型的层组件现在可在现有的生物序列深度学习Basenji代码库中获取:https://github.com/calico/basenji,同样采用开源Apache 2.0许可证。
预训练的Enformer模型可在TF-Hub上获取,使用者可以轻松地在新数据上运行它:https://tfhub.dev/deepmind/enformer/1。我们还计划在Kipoi模型库中发布它。我们提供代码示例(enformer-usage.ipynb)说明如何使用该模型对遗传变异进行评分。
最后,我们在这里提供了1000个基因组队列中所有常见变异(在任何人群中MAF>0.5%)的变异效应预测,采用开放的创作共用CC-BY 4.0许可证。为了使这些预测更易获取,我们使用PCA将5313个特征提炼为20个高度信息量的变异得分,以保持发布文件大小可控(总共10M个变异小于1GB,而不是100GB),同时保持较高的预测准确性(使用所有特征的GTEx精细定位分类auROC为0.747,而使用提炼后的特征为0.743)。
扩展图
扩展数据图1
扩展数据图2
扩展数据图3
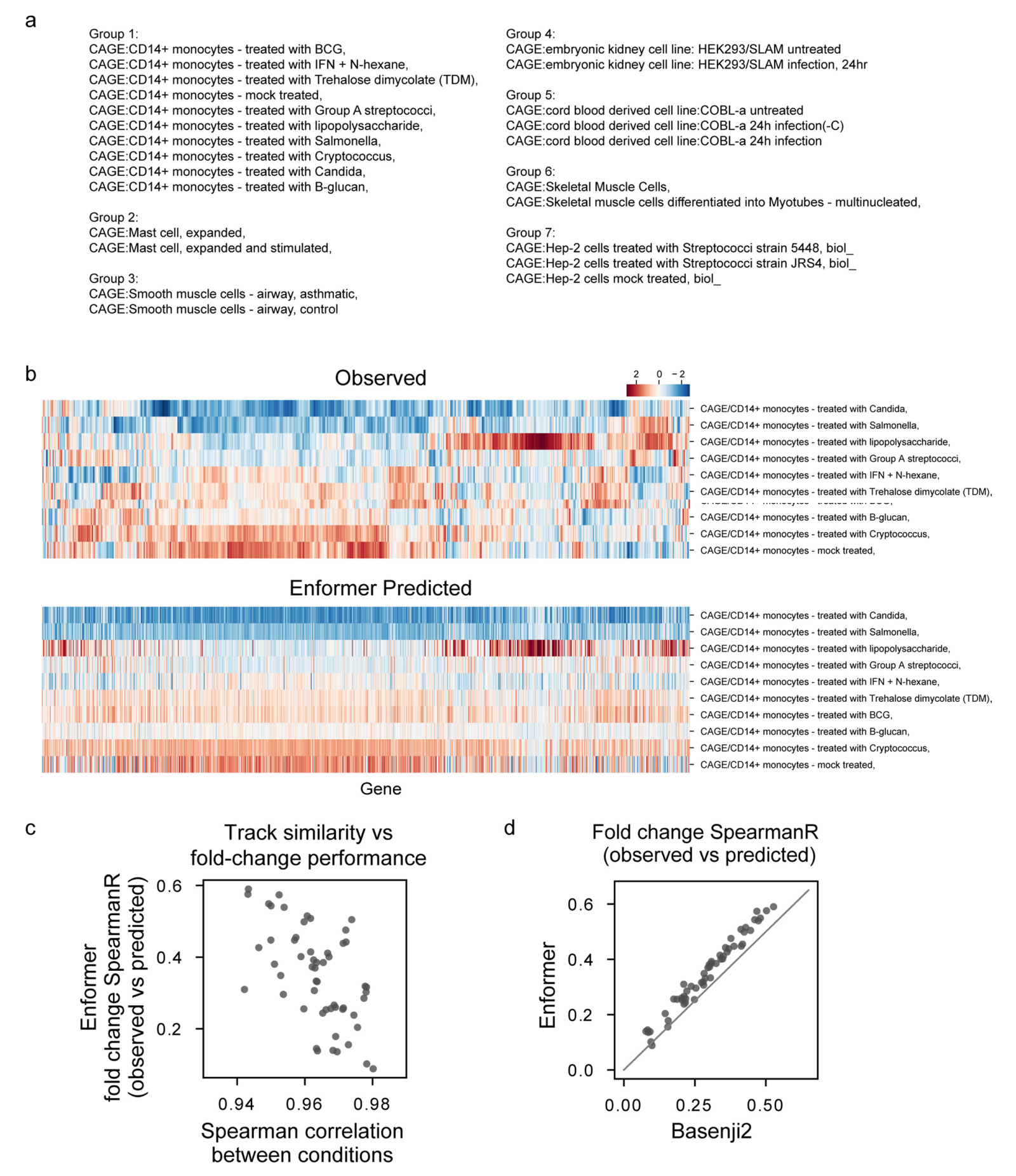
扩展数据图4
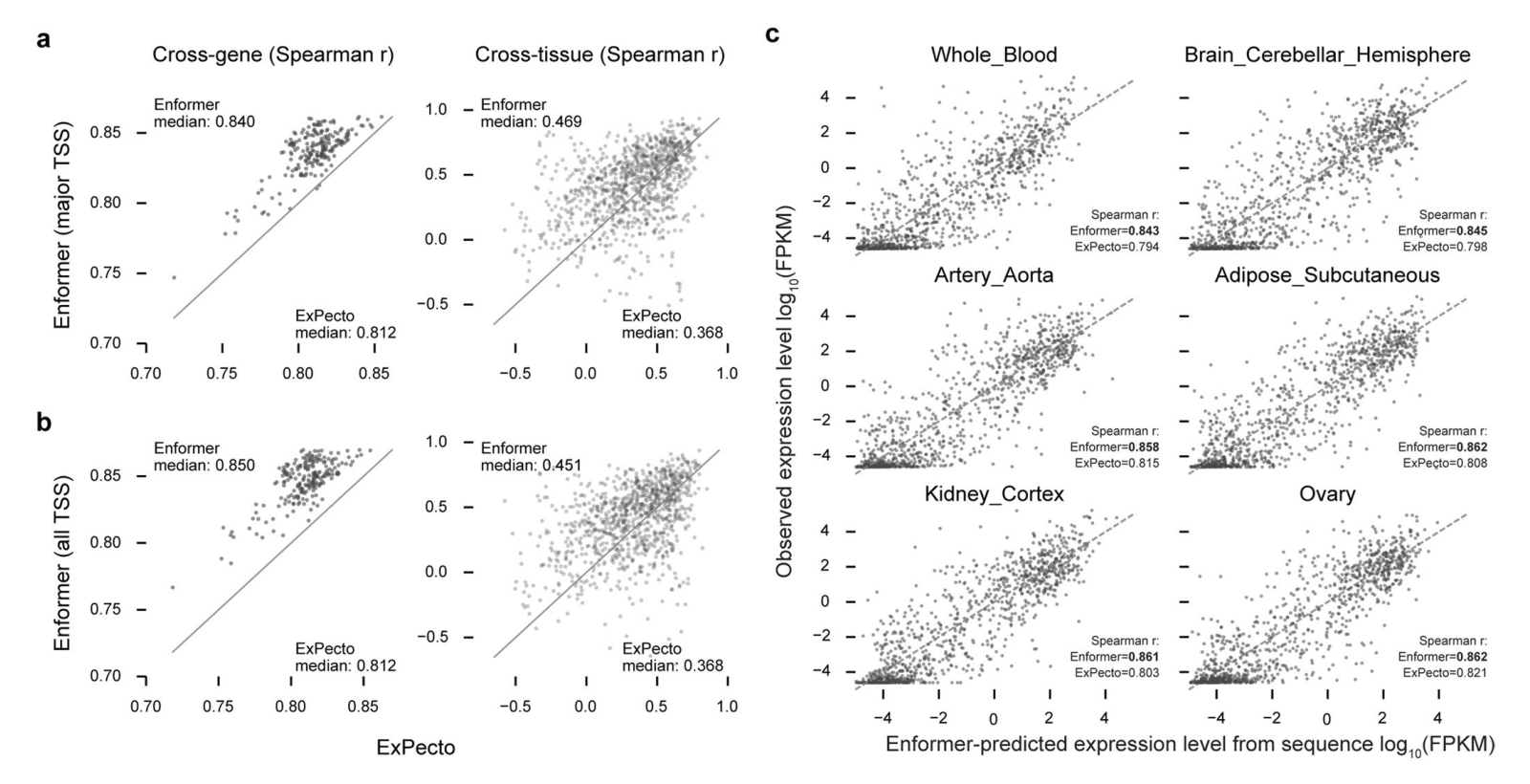
扩展数据图5
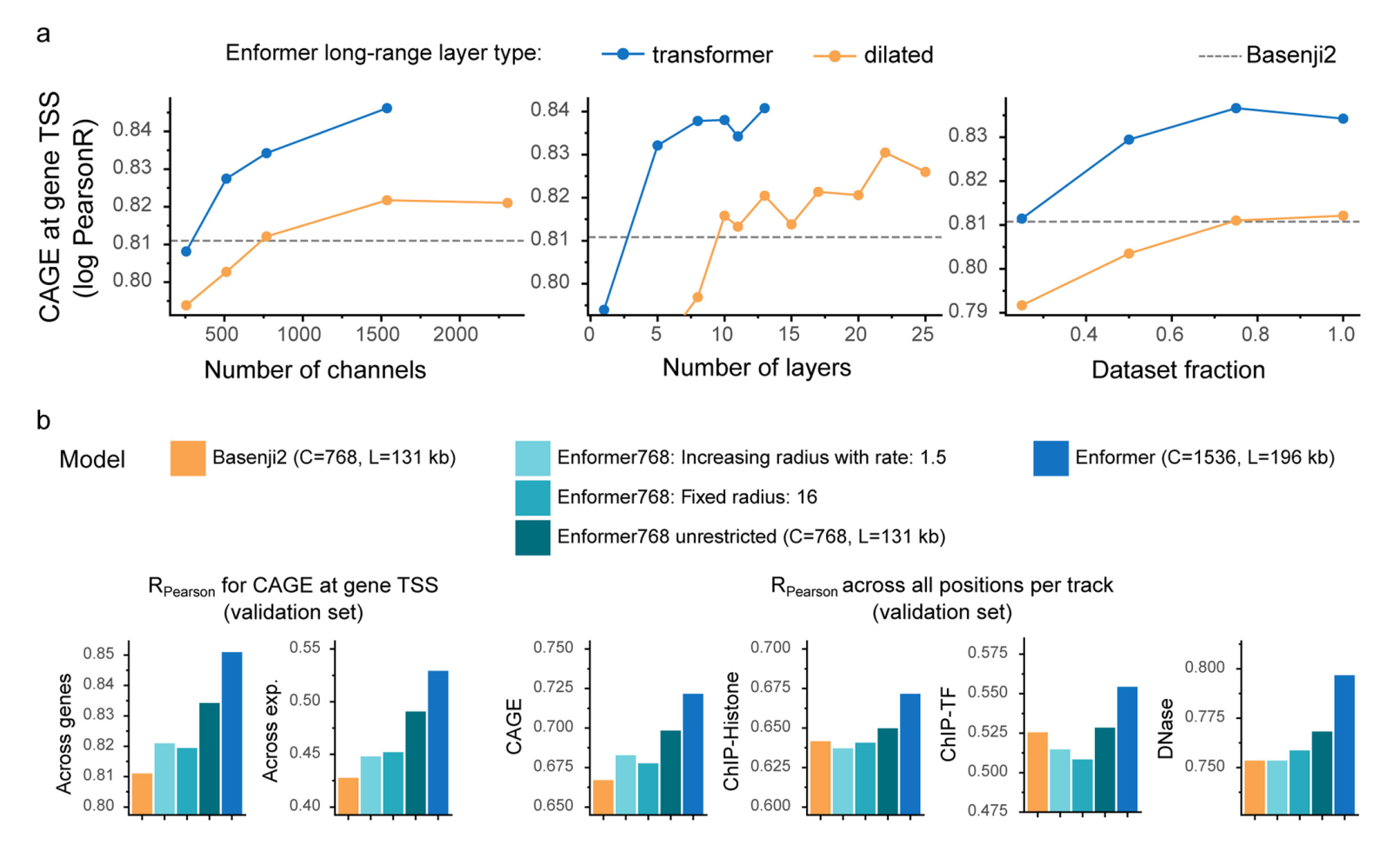
扩展数据图6
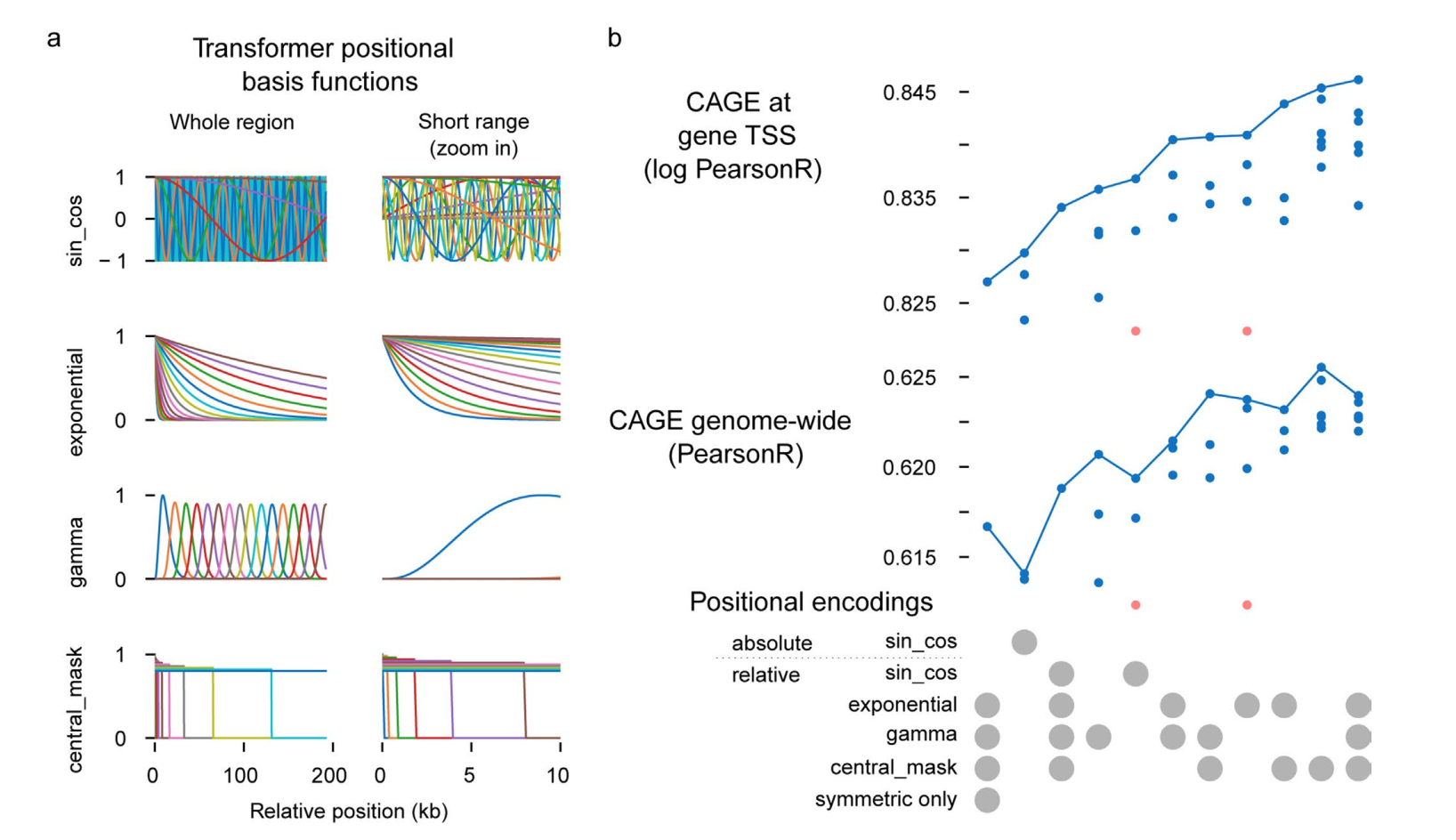
扩展数据图7
扩展数据图8
扩展数据图9
扩展数据图10
参考文献
-
Zhou, J. et al. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. 50, 1171-1179 (2018).
-
Kelley, D. R. Cross-species regulatory sequence activity prediction. PLoS Comput. Biol. 16, e1008050 (2020).
-
Kelley, D. R. et al. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res. 28, 739-750 (2018).
-
Agarwal, V. & Shendure, J. Predicting mRNA abundance directly from genomic sequence using deep convolutional neural networks. Cell Rep. 31, 107663 (2020).
-
Gasperini, M., Tome, J. M. & Shendure, J. Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet. 21, 292-310 (2020).
-
Vaswani, A. et al. Attention is all you need, in Advances in Neural Information Processing Systems 5998-6008 (2017).
-
Brown, T. B. et al. Language models are few-shot learners. in Advances in Neural Information Processing Systems (2020).
-
Ji, Y., Zhou, Z., Liu, H. & Davuluri, R. V. DNABERT: pre-trained bidirectional encoder representations from transformers model for DNA-language in genome. Bioinformatics https://doi.org/10.1093/bioinformatics/btab083 (2021).
-
Gasperini, M. et al. A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell 176, 377-390 (2019).
-
FANTOM Consortium and the RIKEN PMI and CLST (DGT). A promoter-level mammalian expression atlas. Nature 507, 462-470 (2014).
-
Heinz, S., Romanoski, C. E., Benner, C. & Glass, C. K. The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 16, 144-154 (2015).
-
Shrikumar, A., Greenside, P. & Kundaje, A. Learning important features through propagating activation differences. in International Conference on Machine Learning 3145-3153 (PMLR, 2017).
-
Fulco, C. P. et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664-1669 (2019).
-
Eraslan, G., Avsec, Ž., Gagneur, J. & Theis, F. J. Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet. 20, 389-403 (2019).
-
Avsec, Ž. et al. Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet. https://doi.org/10.1038/s41588-021-00782-6 (2021).
-
ENCODE Project Consortium et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699-710 (2020).
-
Ghandi, M. et al. gkmSVM: an R package for gapped-kmer SVM. Bioinformatics 32, 2205-2207 (2016).
-
Zhou, J. & Troyanskaya, O. G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 12, 931-934 (2015).
-
Kelley, D. R., Snoek, J. & Rinn, J. L. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 26, 990-999 (2016).
-
Consortium, T. G., The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318-1330 (2020).
-
Reshef, Y. A. et al. Detecting genome-wide directional effects of transcription factor binding on polygenic disease risk. Nat. Genet. 50, 1483-1493 (2018).
-
Wang, Q. S. et al. Leveraging supervised learning for functionally informed fine-mapping of cis-eQTLs identifies an additional 20,913 putative causal eQTLs. Nat. Commun. 12, 3394 (2021).
-
Wang, G., Sarkar, A., Carbonetto, P. & Stephens, M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. 82, 1273-1300 (2020).
-
Weissbrod, O. et al. Functionally informed fine-mapping and polygenic localization of complex trait heritability. Nat. Genet. 52, 1355-1363 (2020).
-
Kircher, M., Xiong, C., Martin, B. & Schubach, M. Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nature 10, 3583 (2019).
-
Shigaki, D. et al. Integration of multiple epigenomic marks improves prediction of variant impact in saturation mutagenesis reporter assay. Hum. Mutat. 40, 1280-1291 (2019).
-
Lee, D. et al. A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet. 47, 955-961 (2015).
-
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886-D894 (2019).
-
Alipanahi, B., Delong, A., Weirauch, M. T. & Frey, B. J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 33, 831-838 (2015).
-
Villar, D. et al. Enhancer evolution across 20 mammalian species. Cell 160, 554 (2015).
-
Linder, J. et al. A generative neural network for maximizing fitness and diversity of synthetic DNA and protein sequences. Cell Syst. 11, 49-62 (2020).
-
Fudenberg, G., Kelley, D. R. & Pollard, K. S. Predicting 3D genome folding from DNA sequence with Akita. Nat. Methods 17, 1111-1117 (2020).
-
Schwessinger, R. et al. DeepC: predicting 3D genome folding using megabase-scale transfer learning. Nat. Methods 17, 1118-1124 (2020).
-
Schreiber, J., Durham, T., Bilmes, J. & Noble, W. S. Avocado: a multi-scale deep tensor factorization method learns a latent representation of the human epigenome. Genome Biol. 21, 81 (2020).
-
Nair, S., Kim, D. S., Perricone, J. & Kundaje, A. Integrating regulatory DNA sequence and gene expression to predict genome-wide chromatin accessibility across cellular contexts. Bioinformatics 35, i108-i116 (2019).
-
Tay, Y., Dehghani, M., Bahri, D. & Metzler, D. Efficient transformers: a survey. Preprint at https://arxiv.org/abs/2009.06732 (2020).
-
Richter, F. et al. Genomic analyses implicate noncoding de novo variants in congenital heart disease. Nat. Genet. 52, 769-777 (2020).
-
Zhou, J. et al. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat. Genet. 51, 973-980 (2019).
-
Gupta, S., Stamatoyannpoulos, J. A., Bailey, T. L. & Noble, W. Quantifying similarity between motifs. Genome Biology 8, R24 (2007).
-
Shaw, P., Uszkoreit, J. & Vaswani, A. Self-attention with relative position representations. In Proceedings of the 2018 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 2 (Short Papers) 464-468 (2018).
-
Dai, Z. et al. Transformer-XL: Attentive language models beyond a fixed-length context. In Proceedings of the 57th Annual Meeting of the Association for Computational Linguistics 2978-2988 (2019).
-
Kent, W. J. The Human Genome Browser at UCSC. Genome Research 12, 996-1006 (2002).
-
Reynolds, M. et al. Open sourcing Sonnet — a new library for constructing neural networks. https://deepmind.com/blog/open-sourcing-sonnet (2017).
-
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825-2830 (2011).
-
Klein, J. C. et al. A systematic evaluation of the design and context dependencies of massively parallel reporter assays. Nat. Methods 17, 1083-1091 (2020).
-
Avsec, Žiga et al. Enformer (Version 3.0) (Zenodo, 2021); https://doi.org/10.5281/zenodo.5098375
-
Avsec, Ž. et al. The Kipoi repository accelerates community exchange and reuse of predictive models for genomics. Nat. Biotechnol. 37, 592-600 (2019).
