
文章链接
Dalla-Torre, H., Gonzalez, L., Mendoza-Revilla, J. et al. Nucleotide Transformer: building and evaluating robust foundation models for human genomics. Nat Methods (2024). https://doi.org/10.1038/s41592-024-02523-z
简介
Nucleotide Transformer:提出了一个构建和评估人类基因组学的稳健基础模型
摘要
从DNA序列预测分子表型一直是基因组学中的一个长期挑战,这主要受限于标注数据的数量以及在不同任务间迁移学习的困难。在此,我们展示了一项关于在DNA序列上预训练的基础模型(称为Nucleotide Transformer)的广泛研究。这些模型参数规模从5000万到25亿不等,整合了来自3,202个人类基因组和850个不同物种基因组的信息。这些转换器模型能够产生特定上下文的核苷酸序列表征,使其即使在低数据量情况下也能进行准确预测。我们展示了这些开发的模型可以以低成本微调来解决各种基因组学应用。尽管没有监督,这些模型学会了将注意力集中在关键基因组元件上,并可用于改进遗传变异的优先级排序。在基因组学中训练和应用基础模型为从DNA序列进行准确的分子表型预测提供了一种广泛适用的方法。
分子表型:基因表达在分子水平上的具体体现,包括细胞内的蛋白质、RNA、代谢物等分子及其相互作用网络所呈现的可测量特征,它是连接基因型和传统表型的桥梁,帮助我们理解基因如何通过分子机制影响生物体的性状表现。
创新点
- 规模和多样性的突破:
- 构建了迄今为止最大的
DNA基础模型之一,参数规模达到25亿 - 使用了迄今最大规模和最多样化的训练数据集,包含
3,202个人类基因组和850个不同物种的基因组数据
- 技术创新:
- 提出了
NT-v2优化版本,通过架构改进实现了模型小型化(参数减少50倍)的同时保持或提升性能 - 创新性地采用了参数高效的微调技术(
IA3),仅需调整0.1%的参数就能实现高性能
- 应用创新:
- 无需监督即可识别关键基因组元素
- 提出了基于
零样本的遗传变异评分方法 - 在多个具有挑战性的基因组学任务上达到或超过了专门设计的监督学习模型的性能
主要内容
读前须知
- 论文解读尽可能的还原原文,若有不恰当之处,还请见谅;
- 排版上,插图会尽量贴近出处,而
补充图表均在文末“补充信息”的下载链接中; - 左边👈有目录,可自行跳转至想看的部分;
- 部分专业术语翻译成中文可能不太恰当,此时会用括号标明它的英文原文,如感受野(
Receptive field)。请注意,仅首次出现会标明;
引言
人工智能(AI)中的基础模型的特点是其大规模性质,包含数百万经过大量数据集训练的参数。这些模型可以适应广泛的后续预测任务,并且已经深刻地改变了AI领域。自然语言处理(NLP)中的著名例子包括所谓的语言模型(LM)BERT和GPT。近年来LM变得越来越受欢迎,这要归功于它们能够在未标记的数据上进行训练,从而创建能够解决下游任务的通用表示。它们实现全面语言理解的一个方法是通过解决数十亿次完形测试,在这些测试中它们预测给定句子中空白处的正确单词。这种方法被称为掩码语言建模。
掩码:深度学习中是一种二进制标记机制,通常用0和1表示需要忽略或保留的数据。它可以用于处理缺失值、控制注意力机制的关注范围、实现序列填充对齐等任务。在Transformer中,掩码尤其重要,它既可以防止模型在预测时看到未来信息(自回归掩码),也可以帮助处理不等长序列(填充掩码)。
早期将这一目标应用于生物学的基础模型涉及在蛋白质序列上训练LM,它们的任务是在大型蛋白质序列数据集中预测被掩码的氨基酸。这些蛋白质LM在应用于下游任务时通过迁移学习,展示了与之前的方法竞争甚至超越之前方法的能力,例如预测蛋白质结构和功能,即使在数据稀缺的情况下也是如此。
除了蛋白质序列之外,DNA序列中编码的依赖模式在理解基因组过程中起着基础性作用,从表征调控区域到评估单个变异在其单倍型环境中的影响。在这种情况下,专门的深度学习(DL)模型已经被训练来揭示DNA中有意义的模式。例如,DL模型已被用于从DNA序列预测基因表达,最近的进展将卷积神经网络和transformer架构相结合,能够编码位于上游100千碱基(kb)的调控元件。
现代基因组学研究产生的大量数据既是机遇也是挑战。一方面,物种和群体之间的自然变异复杂模式可以轻易获得;另一方面,需要强大的DL方法来处理大规模数据,以便从未标记的数据集中准确提取信号。在基因组学中训练的大型基础模型似乎是值得探索的方法来应对这一挑战。
在这里,我们构建了用于编码基因组序列的稳健基础模型,称为Nucleotide Transformer(NT),并提供了系统性研究和基准测试来评估其性能。我们通过构建四个不同大小的LM开始研究,从5亿到25亿参数不等。这些模型在三个不同数据集上进行预训练,包括人类参考基因组、3,202个多样化人类基因组的集合,以及来自各种物种的850个基因组。
训练之后,我们以两种方式利用每个模型的表示(嵌入)。为了评估NT在适应各种任务时的性能稳定性,我们在一组18个精选的基因组预测任务上训练了每个模型,并将它们与三个替代的DNA基础模型以及一个最先进的非基础模型进行比较,使用系统的十折交叉验证程序。
十折交叉验证:一种数据集划分方法,将数据集平均分成10份,每次用9份作为训练集,剩下1份作为验证集,轮流进行10次,最终取10次评估结果的平均值作为模型性能的估计。
此外,为了扩大我们的评估,我们将我们表现最好的模型与三个针对特定任务优化的最先进的监督基线模型进行了比较。为了破译在预训练期间学习的序列特征,我们探索了模型的注意力图和复杂度(perplexity),并对其嵌入进行了数据降维。
此外,我们还评估了嵌入对人类功能重要遗传变异影响进行建模的能力,通过零样本评分。基于初始实验的发现,我们开发了第二组四个LM,参数从5亿减少到5千万,以研究这类模型的缩放规律。我们成功构建了一个模型,仅用十分之一的参数就达到了之前最佳模型的性能,同时将感受野大小翻了一番。
感受野:神经网络中某一层的神经元能够"看到"的输入区域范围。
结果
Nucleotide Transformer模型准确预测基因组学任务
我们开发了一系列基于transformer的DNA语言模型(NT),它们从6kb未注释的基因组数据中学习了通用的核苷酸序列表示(图1a和方法)。受NLP中的趋势启发,即更大的训练数据集和模型规模已经显示出改进的性能,我们构建了具有不同参数规模和数据集的transformer模型:
-
在人类参考基因组序列上训练的
5亿参数模型("Human ref 500M") -
一个
5亿参数模型("1000G 500M") -
一个
25亿参数模型("1000G 2.5B")(它们都在3,202个遗传多样性的人类基因组上训练) -
一个
25亿参数模型,包含来自不同门类的850个物种("Multispecies 2.5B"),其中包括11个模式生物(图1c和补充表1-4)。
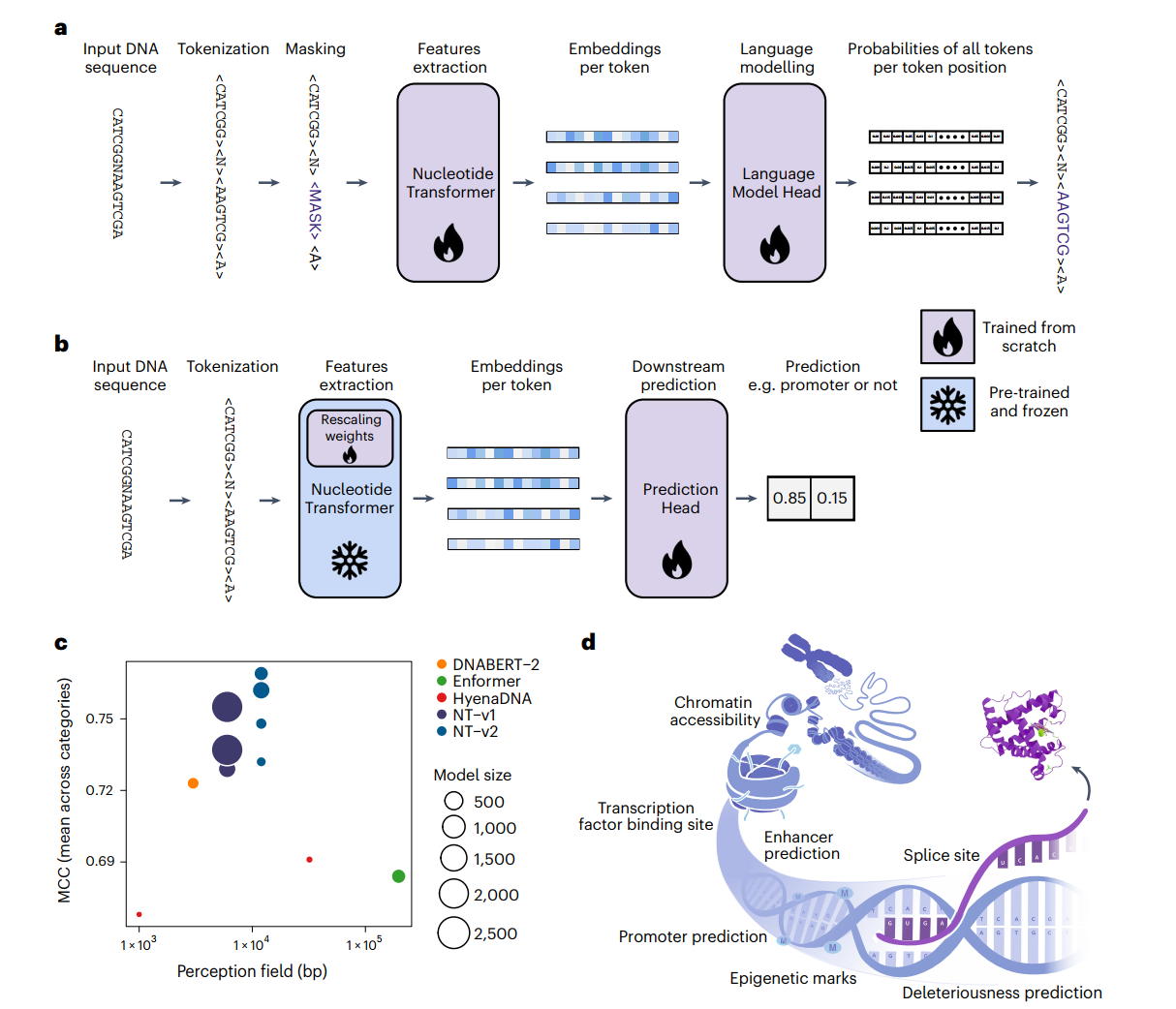
为了评估这些模型在预测各种分子表型方面的效果,我们从公开可获得的资源中整理了18个基因组数据集,包括剪接位点预测任务(GENCODE)、启动子任务(Eukaryotic Promoter Database)以及组蛋白修饰和增强子任务(ENCODE),每个数据集的大小都经过合理设计,以实现快速和严格的交叉验证程序(图1d,补充表5和方法)。
虽然对监督模型来说有更大的数据集可用,但这18个基因组数据集的编译提供了多样化和稳健的选择,用于严格检验模型在各种任务中的适应性,并与其他DNA自监督基础模型进行比较。这些数据集被处理成标准化格式以方便实验并确保在评估大型语言模型性能时的可重复性(方法)。我们通过两种不同的技术评估了我们的transformer模型:探测和微调(图1b)。
探测是指使用学习到的语言模型DNA序列嵌入作为输入特征,输入到更简单的模型中来预测基因组标签。具体来说,我们使用逻辑回归或一个最多包含两个隐藏层的小型多层感知器(MLP),探测了语言模型的十个任意选择的层。在微调的情况下,语言模型的头部被替换为分类或回归头部,并使用参数高效的技术进行再训练(方法)。为了确保不同模型之间的公平和准确比较,我们实施了十折交叉验证策略。
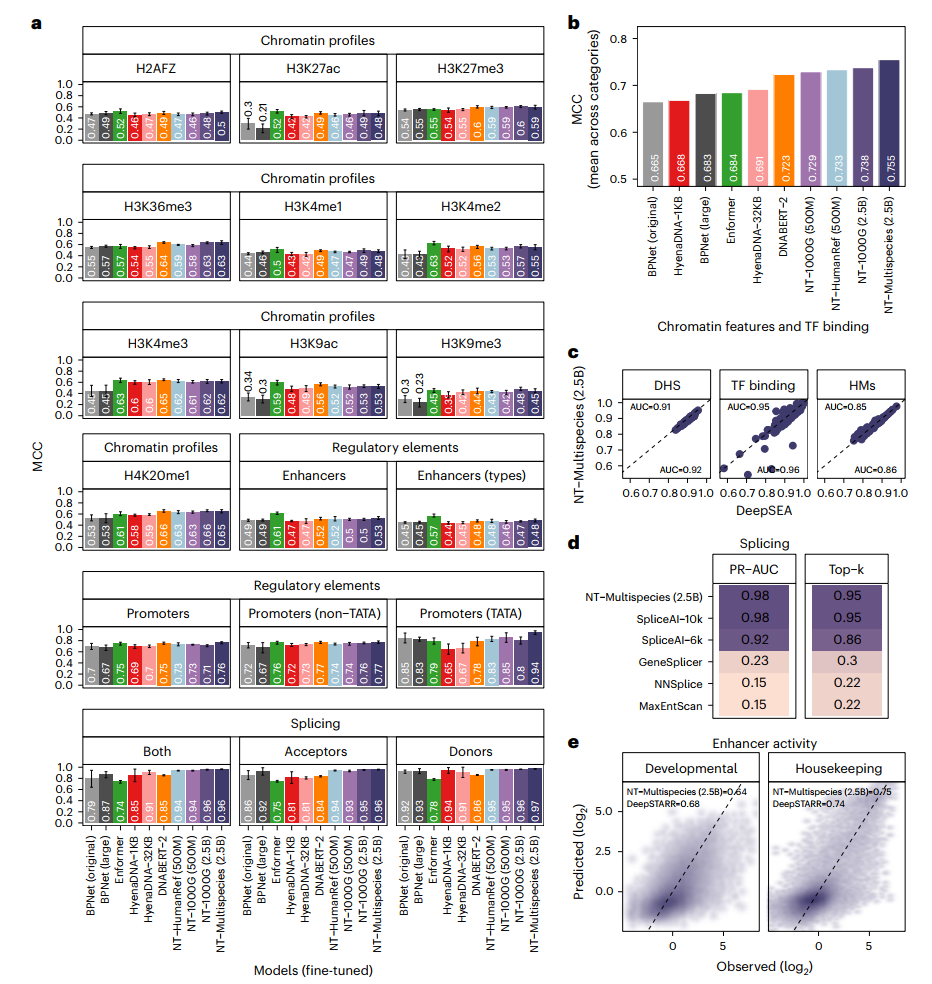
为了将我们的预训练基础模型方案与该领域的标准监督方法进行比较,我们从头开始在18个任务中的每一个上训练了不同版本的BPNet卷积架构(方法)。BPNet架构在基因组学中得到了广泛应用,代表了一个非常强大的默认架构,用于通过监督学习从头开始建模小型数据集。我们观察到原始BPNet模型在各个任务中都表现出强劲的性能(平均Matthews相关系数(MCC)为0.665),通过将其规模增加到2800万参数,我们可以将其提高(平均MCC为0.683),这证实了直接监督的卷积架构在基因组学任务中表现得非常好(图2a,b)。接下来,我们评估了NT模型的探测和微调与我们基准数据集上的这些监督基线模型相比如何。如果所得的两个标准差重叠或优于报告的基线值,我们认为这些模型是等同于或优于其他模型的。
BPNet:一个用于预测DNA序列与蛋白质结合的深度学习架构,它结合了扩张卷积网络和基于峰值的损失函数。该网络首先使用多尺度的扩张卷积来捕获DNA序列特征,然后通过解码器预测结合位点的概率分布,最后采用特殊的损失函数来优化对结合峰值的预测。BPNet的一个重要特点是可以生成序列重要性分数,帮助解释模型的预测结果。
使用这个标准,NT模型仅通过探测就在5个任务中匹配基线BPNet模型,并在18个任务中的8个任务中超越了它们(补充图1和补充表6),并显著优于来自原始标记的探测。与最近的工作一致,我们观察到最佳性能既取决于模型也取决于层(补充表8)。我们还注意到,最高的模型性能从来不是通过使用最后一层的嵌入获得的,这与早期工作一致。例如,在增强子类型预测任务中,我们观察到性能最高和最低的层之间的相对差异高达38%,表明在各层之间学习到的表示有显著差异(补充图3)。与我们的探测策略相比,我们的微调模型在18个基线模型中要么匹配(n=6)要么超越(n=12)(图2a,b和补充表7和9)。值得注意的是,微调的NT模型优于探测模型,而且更大和更多样化的模型持续优于较小的模型。这些结果支持了有必要针对特定任务微调NT基础模型以实现更好的性能。
我们的结果还表明,在多样化数据集上训练的Multispecies 2.5B模型在几个基于人类测试的任务上优于或匹配1000G 2.5B模型(图2a,b)。这意味着增加序列多样性的策略,而不仅仅是增加模型规模,可能会带来更好的预测性能,特别是在计算资源有限的情况下。
微调在之前的工作中没有被广泛探索,可能是由于其计算需求较高。我们通过采用最新的参数高效微调技术克服了这一限制,该技术仅需要总模型参数的0.1%(图1b和方法)。这种方法允许在单个GPU上更快地进行微调,比所有微调参数减少了1,000倍的存储需求,同时仍能提供相当的性能。在实践中,我们观察到即使使用简单的下游模型对嵌入进行探测,严格的探测也比微调更慢且计算强度更大。这种差异源于诸如层的选择、下游模型选择和超参数对性能的重要影响。此外,微调表现出更小的性能方差,增强了该方法的稳健性。总的来说,这种通用方法是多功能的,无需调整模型架构或超参数即可适应各种任务。这与监督模型形成鲜明对比,后者通常具有不同的架构,并且需要为每个任务从头开始训练。
最后,我们旨在评估大型DNA语言模型与使用大量数据集训练并优化架构的稳健基线进行竞争的潜力。为此,我们将Multispecies 2.5B模型应用于另外三个基因组预测任务,这些任务包括:
- 对来自多样化人类细胞和组织的
919个染色质谱的分类 - 预测整个人类基因组中的规范剪接受体和供体位点
- 预测来自果蝇
S2细胞的发育和管家增强子活性(见方法)
剪接位点:真核生物基因中内含子和外显子连接处的特定序列,主要包括5’剪接位点(供体位点)和3’剪接位点(受体位点)。在RNA前体剪接过程中,剪接体通过识别这些保守序列位点来确定剪接位置,将内含子去除并将外显子连接起来。准确的剪接位点识别对于正确的基因表达和蛋白质合成至关重要。
值得注意的是,尽管没有对其原始微调架构进行额外的更改或优化,Multispecies 2.5B模型达到了与专门化DL模型紧密一致的性能水平。例如,在分类染色质特征谱的情况下,我们获得的曲线下面积(AUC)值平均仅比DeepSEA实现的值低约~1%(图2c)。
关于预测pre-mRNA转录本中的每个位置是否是剪接供体、剪接受体或都不是,我们调整了NT模型以提供核苷酸级别的剪接位点预测,并实现了95%的top-k准确率和0.98的精确率-召回率AUC(图2d)。值得注意的是,我们的2.5B 6-kb上下文模型与最先进的SpliceAI-10k的性能相匹配,后者在15-kb输入序列上训练,此外还优于其他剪接基线;并且在测试6-kb输入序列时优于SpliceAI。
最后,在管家和发育增强子预测的情况下,我们的模型略微超过(1%)和获得较低(4%)的相关值(图2e),与DeepSTARR相比。在这三个不同任务中,我们还对我们的参数高效微调和全模型微调(训练模型的所有参数以优化其在特定任务或数据集上的性能)进行了比较。
值得注意的是,在染色质和剪接预测方面我们没有观察到显著改进,在增强子活性预测方面仅有适度的3%提升(补充图2),支持我们使用高效微调方法。总的来说,我们广泛的基准测试和结果展示了NT作为一种通用方法在处理许多不同基因组学任务时的灵活性和高准确性。
基因组学基础模型的基准测试
我们将NT模型与其他基因组学基础模型进行了比较:DNABERT-2、HyenaDNA(1-kb和32-kb上下文长度)和Enformer(将其用作预训练模型的替代架构)(图2a,b和方法)。我们将DNABERT-1排除在这个比较之外,因为它只能处理最大512 bp的输入长度,因此不能用于大多数任务。为确保公平比较,所有模型都使用相同的协议在18个下游任务中进行微调和评估(方法)。
与DNABERT-2、HyenaDNA-32-kb和Enformer相比,我们的Multispecies 2.5B模型在所有任务中达到了最高的总体性能(图2a,b和表9)。尽管如此,Enformer在增强子预测和一些染色质任务中取得了最佳性能,证明它可以成为一个强大的DNA基础模型。我们的模型在所有启动子和剪接任务中都表现优于其他每个模型。值得注意的是,尽管HyenaDNA在人类参考基因组上进行预训练,我们的Multispecies 2.5B模型在所有18个任务中要么匹配(n=7)要么超越(n=11)它,突显了在多样化基因组序列集上预训练的优势。我们建立了一个包含所有模型在每个任务上结果的交互式排行榜(https://huggingface.co/spaces/InstaDeepAI/nucleotide_transformer_benchmark)以方便比较。这代表了基因组学基础模型的广泛基准测试,应该作为进一步发展基因组学中语言模型的参考(图1c)。
以无监督的方式检测已知的基因组元件
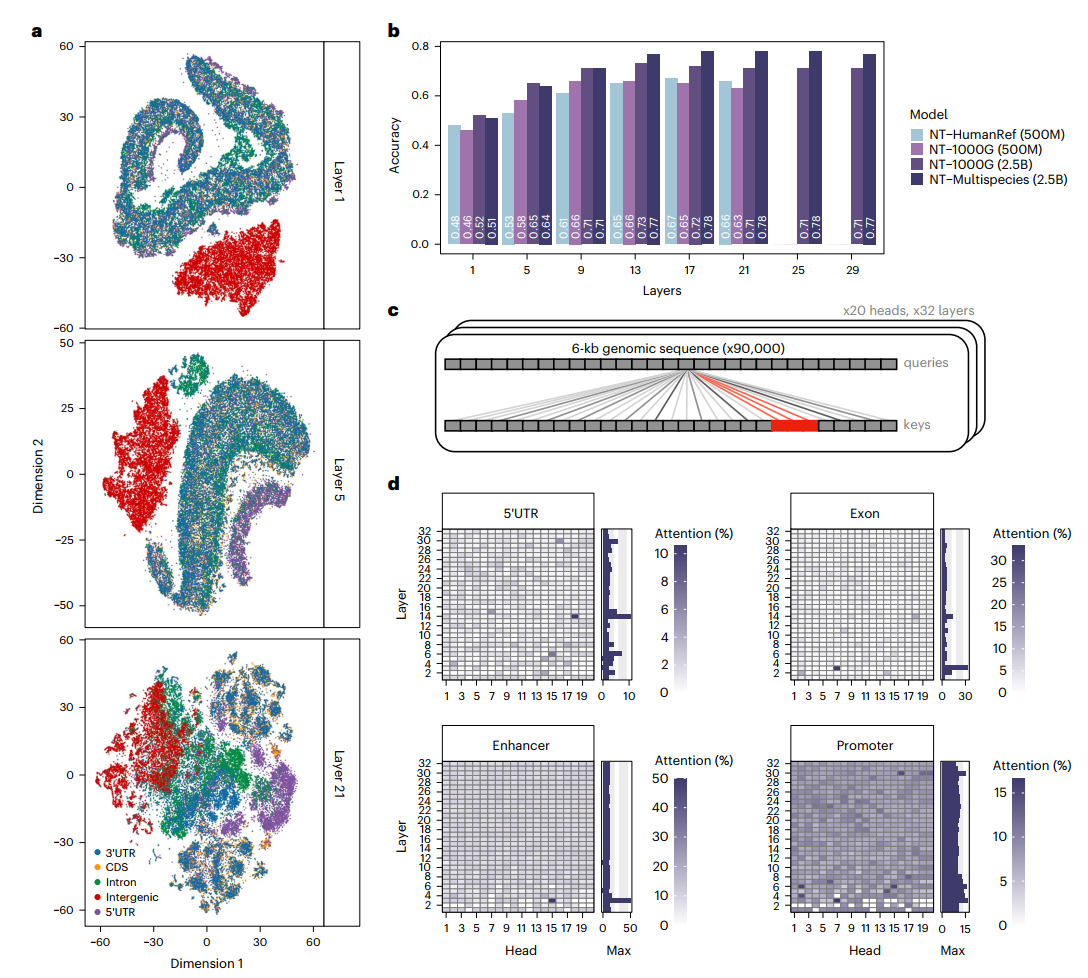
为了深入了解NT模型的可解释性,并理解这些模型在进行预测时使用的序列元素类型,我们探索了它们架构的不同方面。首先,我们评估了这些嵌入在多大程度上可以捕获与五种基因组元件相关的序列信息(补充表7和10)。我们观察到,NT模型在没有任何监督的情况下,学会了区分具有独特注释的基因组序列,包括基因间区、内含子、编码区和非翻译区(UTRs),尽管在不同层次上的熟练程度不同(图3a、补充图7和方法)。特别是,500M大小的模型和那些在较少多样性序列上训练的模型,在基因组区域之间表现出较低的区分度,这再次证实了最大的模型在自监督训练期间更能捕获相关基因组模式的能力。
在Multispecies 2.5B模型的情况下,第1层观察到基因间和基因区域之间最强的区分,其次是第5层的5'UTR区域,以及第21层大多数区域之间的区分(图3a)。3'UTR区域与其他元素的有限区分表明,该模型尚未完全学会区分这种类型的元素,或者如先前研究所建议的,这些区域中可能有许多注释错误。与这些观察结果一致,我们的监督探测策略展示了对这些元素的高分类性能,特别是在更深层次,准确率值超过0.78(图3b)。这证明了NT模型已经以无监督的方式学会在其嵌入中检测已知的基因组元素,这可以被用于高效的下游基因组任务预测。
接下来,我们通过注意力机制的视角对模型进行分析,以理解注意力层捕获和使用了哪些序列区域。我们计算了每个模型头和层对包含九种不同类型基因组元素(与基因结构和调控特征相关)的序列的注意力百分比(图3c)。
注意力机制:一种让模型能够动态关注输入中重要部分的计算方法。它通过计算查询(query)与键值对(key-value pairs)的相关性来分配权重,从而让模型对重要信息给予更多"关注"。在Transformer中,自注意力机制让序列中的每个位置都能与其他位置进行交互,捕获长距离依赖关系,已成为现代深度学习的核心组件之一。
transformer模型中的相关概念可参考这篇文章:
从形式上讲,当注意力头的注意力百分比显著超过该元素在预训练数据集中自然出现的频率时,就认为该注意力头识别出了特定元素(方法)。例如,50%的比例意味着,在人类基因组中平均而言,该特定头部50%的注意力指向了感兴趣的元素类型。通过将这种方法应用于在6kb序列中不同位置可能出现的每种类型元素(约10,000个不同序列,其中元素占序列的2-11%;补充表10),我们发现注意力在其不同的头部和层中明显聚焦于特定类型的基因组元素(图3d和补充图8-16)。
跨层的显著注意力头数量在不同模型间有显著差异,其中Multispecies 2.5B模型对内含子(640个头中的117个)、外显子(n=72)和转录因子(TF)结合位点(n=74)表现出最多的显著注意力头(补充图8、9和补充表12),尽管包含外显子和TF基序的序列比例相对较小。关于增强子,最大模型的最大注意力百分比最高,例如1000G 2.5B模型实现了接近100%的注意力(补充图15)。对于其他基因组元素,如3'UTR、启动子和TF结合位点,也观察到类似的模式,其中1000G 2.5B模型在首层展示了具有高度注意力的高度专门化头部(补充图8-16)。
为了更深入地了解预训练的NT Multispecies 2.5B模型在更高分辨率下的表现(关注更局部的序列特征),我们研究了不同类型基因组元素的token概率,作为模型学习的序列约束和重要性的度量。具体来说,我们计算了22号染色体上每个6kb窗口中的六聚体token概率(基于每次屏蔽一个token)。我们的发现显示,除了被模型很好重建的重复元素外,预训练模型还学习了各种基因结构和调控元素。这些包括受体和供体剪接位点、polyA信号、CTCF结合位点和其他基因组元素(补充图17a-d)。
polyA信号:真核生物mRNA 3’端的保守序列(通常是AAUAAA),它指导mRNA的切割和多聚腺苷酸化过程,对mRNA的稳定性和成熟至关重要。
CTCF结合位点:CTCF蛋白在基因组上的特异性结合序列,作为绝缘子和染色质结构调控因子,参与基因调控、染色质构象组织和转录绝缘等功能,在基因组三维结构形成中起重要作用。
此外,我们将我们的token预测与MST1R基因外显子11的实验饱和突变剪接实验进行了比较(数据来自Braun等人)。这个分析揭示了实验突变效应与Multispecies 2.5B模型做出的token预测之间存在显著相关性(皮尔逊相关系数(PCC)= 0.44;补充图17e)。该模型不仅捕获了不同剪接位点的约束,还识别出第二个内含子中间对该外显子剪接至关重要的区域。这些结果有力地验证了NT模型在无监督预训练期间获得的生物学知识。
最后,对于在DeepSTARR增强子活性数据上完全微调的Multispecies 2.5B模型,我们检查了它是否特别学习了与增强子活性相关的TF基序及其相对重要性。我们使用了一个数据集,其中包含了数百个增强子序列中五种不同TF基序类型的数百个单独实例的实验突变,并评估了模型在预测这些突变效应方面的准确性。
与最先进的增强子活性DeepSTARR模型相比,我们的模型在四个TF基序上达到了相似的性能,并在Dref基序上展示了更优的性能(补充图18)。总的来说,这些结果说明了NT模型已经获得了恢复基因结构和基因组序列功能特性的能力,并直接将它们整合到其注意力机制中。这种编码的信息应该有助于评估遗传变异的重要性。
预训练的嵌入预测突变的影响
此外,我们评估了NT模型在评估各种遗传变异的严重程度和优先排序具有功能意义的变异方面的能力。我们首先研究了零样本评分的使用,这些评分用于预测模型在训练期间未见过的类别。
具体来说,我们使用嵌入空间中不同方面的向量距离以及从损失函数中得出的评分来计算零样本评分,并比较它们在严重程度不同的十种遗传变异类型中的分布情况(图4a和方法)。令人鼓舞的是,一些零样本评分在模型间表现出与严重程度的中等相关性(补充图19)。这说明了仅通过无监督训练就捕获了与遗传突变潜在严重程度相关的信息,并突出了评估不同评分方法的重要性。评分之间的高度相关性变异也表明,嵌入空间的不同方面可能更有效地捕获与严重程度相关的信息。
在这些评分中,余弦相似度在各个模型中表现出与严重程度最高的相关性, 值范围从-0.35到-0.3(;补充图19)。在所有模型中,最低的余弦相似度评分被分配给影响蛋白质功能的遗传变异,如终止获得变异,以及同义和错义变异(图4b)。相反,我们注意到较高的评分被分配给潜在功能重要性较低的变异,如基因间变异,突出了其在捕获遗传变异严重程度影响方面的潜在用途。
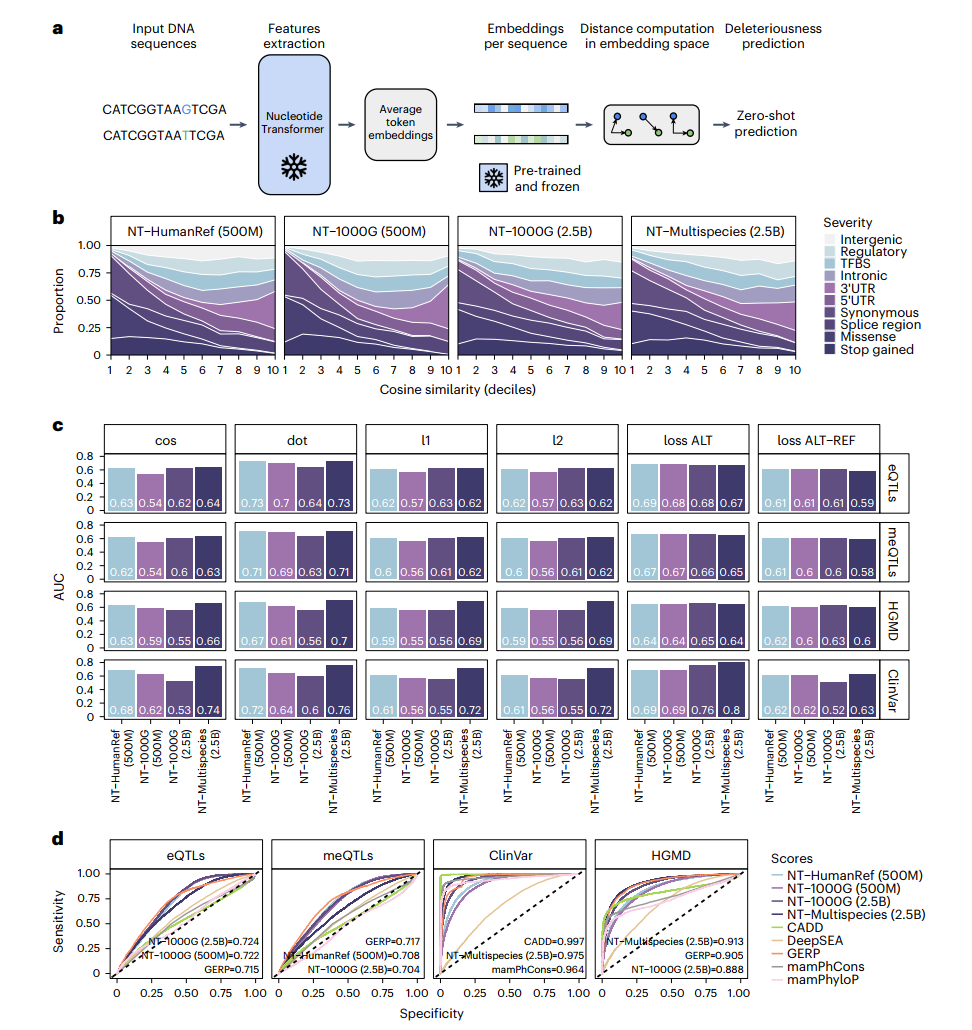
接下来,我们还探索了零样本评分在优先考虑功能变异以及具有致病效应变异方面的潜力。具体来说,我们评估了模型对影响基因表达调控的遗传变异(表达量性状位点,eQTLs)、与DNA甲基化变异相关的遗传变异(甲基化量性状位点,meQTLs)、在ClinVar数据库中注释为致病性的遗传变异以及在人类基因突变数据库(HGMD)中报告的遗传变异进行分类的能力。零样本评分表现出高分类性能,在四个任务中的最高AUC值范围从0.7到0.8(图4c)。在ClinVar变异中获得的最高性能(Multispecies 2.5B模型的AUC为0.80)表明,至少对于高度致病性变异,零样本评分可能可以直接应用。
最后,为了更正式地评估这些模型的有效性,我们还基于微调模型进行预测,并将其性能与多种方法进行了比较。这些方法包括那些测量基因组保守性的方法,以及从在功能特征上训练的模型中获得的评分。值得注意的是,微调模型或略微超过或紧密匹配了其他模型的性能(图4d和补充图20)。
对于优先排序分子表型(eQTLs和meQTLs)表现最好的模型是那些在人类序列上训练的模型,而对于优先排序致病性变异表现最好的模型则是基于多物种序列的模型。考虑到最严重的致病性变异往往由于氨基酸改变而影响基因功能,多物种模型可能利用了跨物种的序列变异来学习位点的保守程度。
我们的结果还表明,对于非编码变异(如eQTLs和meQTLs)的更高预测能力,可以通过从增加的人类遗传变异性中更好地学习序列变异来实现。此外,与零样本评分相比,点积在eQTLs和meQTLs上分别产生了0.73和0.71的AUC值,略微超过或匹配了通过微调模型获得的值。鉴于这些遗传变异往往位于调控区域内,这可能表明这些模型在没有任何监督的情况下,已经学会了区分与基因表达和甲基化变异相关的相关调控基因组特征。这与在层和头部之间观察到的注意力水平一致,特别是对于相关的调控序列,如增强子(图3a)和启动子(补充图13),这些序列已被证明在meQTLs和eQTLs中富集。总的来说,这些结果说明了基于DNA的转换器模型如何能帮助揭示和有助于理解与分子表型和疾病相关的变异的潜在生物学意义。
基因组学中成本效益预测的模型优化
最后,我们探索了通过整合当代架构进展并延长训练时间来优化我们表现最好的模型的可能性。我们开发了四个新的NT模型(NT-v2),参数数量从5千万到5亿不等,并引入了一系列架构改进(补充表1和方法)。这些改进包括引入旋转嵌入、实施swiGLU激活,以及消除MLP偏差和丢弃机制,这与最新的研究相一致。
此外,我们将上下文长度扩展到12kb以容纳更长的序列并捕获更远距离的基因组相互作用。我们将250M和500M参数模型的训练时间延长至包含1万亿个token,这与最近文献中的建议相一致(图5a)。在同一个多物种数据集上预训练后,所有四个NT-v2模型都经过了微调,并在相同的18个下游任务上进行评估,其结果与四个初始NT模型进行了比较(图5b和补充表7、9)。
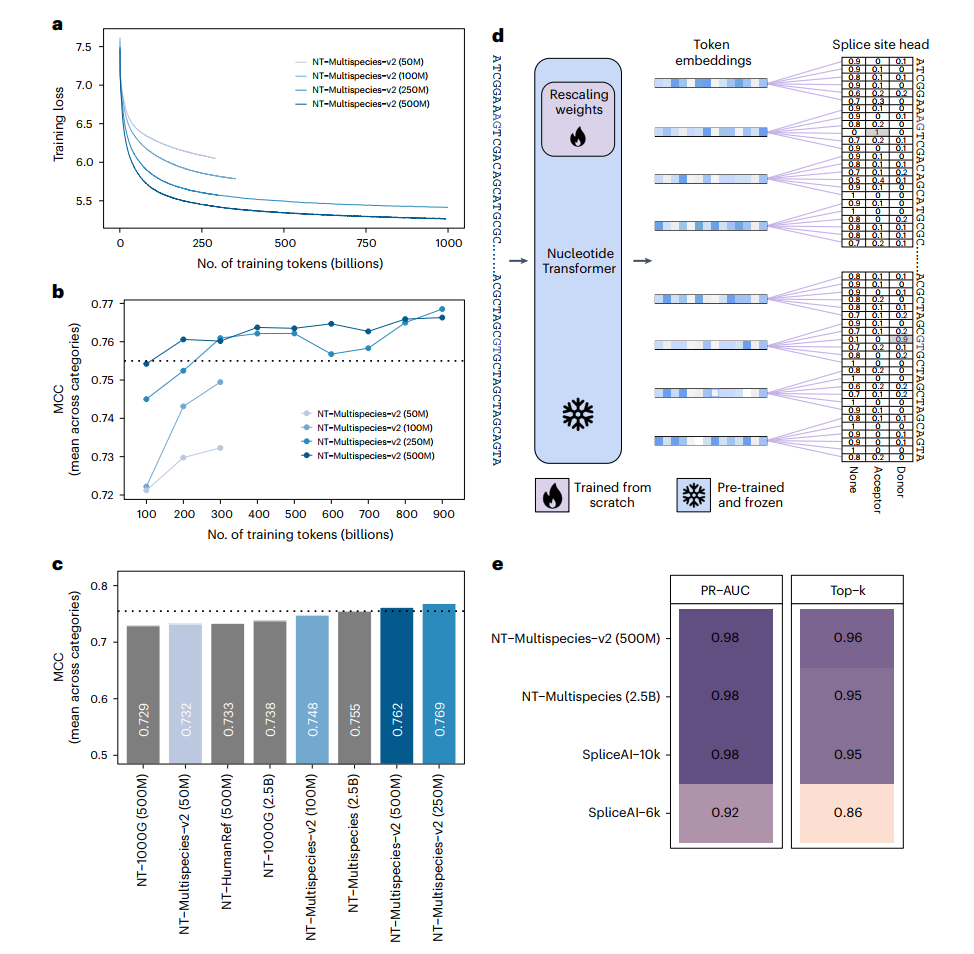
我们观察到50M参数的NT-v2模型实现了与我们两个NT 500M参数模型以及在1000基因组数据集上训练的2.5B参数模型相近的性能。这表明,通过优质的预训练数据集,结合训练技术和架构的进步,可以在显著提高性能的同时实现模型参数数量惊人的50倍减少(图5c)。
事实上,NT-v2 250M参数和500M参数模型在保持参数数量显著较少的同时,成功实现了超越2.5B参数多物种模型的性能,并且将感受野加倍。特别值得注意的是,NT-v2 250M参数模型在我们的基准测试中取得了最佳性能(平均MCC为0.769),同时其参数量仅为2.5B参数模型的十分之一(图5c)。
为了进一步了解更长时间预训练的必要性,我们对NT-v2模型在预训练期间看到的标记数量与性能之间的关系进行了系统性评估(图5b)。这显示了250M参数模型仅在训练达到9000亿个标记后才略微超过500M模型。总的来说,配备了12kb上下文长度的NT-v2模型由于其紧凑的尺寸而适合在经济型加速器上部署。因此,对于希望在下游应用中利用最先进基础模型的用户来说,它们提供了一个经济可行且实用的选择。
作为一个相关的应用案例,在确立了一个表现良好的模型后,我们通过在SpliceAI剪接任务上评估500M模型来评估NT-v2模型更长上下文长度的优势。我们评估了500M模型,因为该模型在剪接相关任务上表现最好(补充表7和9)。我们调整了分类头以预测每个核苷酸位点是剪接供体、受体还是无剪接的概率(图5d和方法)。与NT 6kb模型相比,我们的NT-v2 500M 12kb模型将表现提高了1%,达到了96%的top-k准确率和0.98的精确率-召回率AUC(图5e)。这一性能超过了最先进的SpliceAI-10k模型,后者是在15kb输入序列上训练的。
值得注意的是,我们并没有试图专门针对剪接预测任务优化我们的模型架构;相反,我们采用了与其他下游任务类似的微调方法,仅在分类头上进行调整以产生核苷酸级别的预测。针对特定任务(如剪接)的进一步架构优化很可能会提高性能。总之,这些结果证实了NT v1和v2模型在广泛的基因组学任务中的实用性和有效性,只需要最小的修改和计算能力就能实现高准确率。
讨论
本研究尝试研究在DNA序列上预训练相同规模的transformer模型时,使用不同数据集的影响。基于不同的基因组预测任务的结果表明,种内(在单一物种的多个基因组上训练)和种间(在不同物种的基因组上)变异都显著影响任务的准确性(图1c和2a)。
在考虑的大多数人类预测任务中,在不同物种基因组上训练的模型表现优于仅在人类序列上训练的模型。这表明在不同物种上训练的transformer模型已经学会捕捉到在物种间具有功能重要性的基因组特征,从而能够更好地泛化到各种人类相关的预测任务中。
基于这一发现,我们预计未来的研究可能会受益于利用跨物种的遗传变异性,包括确定采样这种变异性的最佳方式。另一个有趣的研究方向是探索编码种内变异性的不同方法。我们混合所有个体基因组序列的方法只带来了有限的改进,这表明当大多数序列是共享的时候,利用来自不同个体的基因组可能并不那么简单。
本研究中训练的transformer模型参数范围从5000万到25亿,比DNABERT-2大20倍,比Enformer骨干模型大10倍。正如之前在自然语言处理研究中所证明的,我们在18个基因组预测任务上的结果证实,增加模型规模会持续改善性能。为了训练具有最大参数规模的模型,我们在16个计算节点上使用了总共128个GPU,持续28天。
在工程方面投入了大量工作以开发高效的训练程序,充分利用基础设施,这凸显了专门的基础设施和专用软件解决方案的重要性。然而,一旦训练完成,这些模型可以以相对较低的成本进行推理,我们提供了可以将这些模型应用于任何下游任务的笔记本,从而促进进一步的研究。
之前的工作主要基于在生物数据(主要是蛋白质序列)上训练的语言模型,主要通过探测最后一个transformer层来评估下游性能。这种选择可能是由于其使用方便、性能相对较好以及计算复杂度低。在本研究中,我们的目标是通过对不同transformer层、下游模型和超参数进行计算密集型和全面的探测来评估下游准确性。我们观察到最佳探测性能是在中间的transformer层上实现的(补充图1),这与计算生物学领域的最新工作和自然语言处理中的常见实践一致。仅通过探测,Multispecies 2.5B模型在18个任务中的8个任务上超过了BPNet基线模型。
此外,我们探索了一种最新的下游微调技术,该技术在transformer中引入少量可训练权重。这种方法提供了一个相对快速且资源高效的微调程序,与全模型微调(IA3)相比差异很小。值得注意的是,这种微调方法只需要总参数数量的0.1%,使得即使是我们最大的模型也能在单个GPU上在15分钟内完成微调。与大量的探测实验相比,这种技术在使用更少计算资源的同时产生了更好的结果,证实了下游模型工程可以带来性能改进。从操作角度来看,这种技术使得微调在训练和推理方面都能与探测相竞争,同时还能实现更好的性能。
虽然在大型数据集上进行广泛训练的优化架构的监督模型继续展现出优越的性能,但DNA语言方法在包括组蛋白修饰、剪接位点和调控元件表征等多个领域的各种任务中仍具有竞争力。
此外,NT模型代表了一种通用方法,可以无缝地适应人类和非人类物种的各种任务。当处理较小的数据集时,我们的无监督预训练方法的价值变得尤为明显,因为从头开始训练监督模型通常只能产生有限的结果。认识到基础基因组学模型在该领域的关键作用,我们进行了广泛的比较和基准测试研究,将我们的模型与四个具有不同架构的预训练模型进行评估:DNABERT-2、HyenaDNA-1kb、HyenaDNA-32kb和Enformer。这个健全的基准测试将作为未来基因组学领域语言模型发展的参考点。
通过对transformer架构的各种分析,我们已经证明这些模型不仅获得了重建遗传变异的能力(补充说明),还能识别关键的调控基因组元件。这一特性在注意力图、嵌入空间、标记重建和概率分布的分析中得到了证实。所有模型在多个头部和层中都能一致地检测到控制基因表达的关键调控元件,如增强子和启动子。此外,我们观察到每个模型至少包含一层,能产生清晰区分我们分析的五种基因组元件的嵌入。鉴于自监督训练促进了这些元件的检测,我们预计这种方法在未来的研究中可以用于表征或发现新的基因组元件。
我们已经证明,transformer模型可以匹配甚至超越其他用于预测变异效应和致病性的方法。除了开发监督型transformer模型,我们还展示了零样本(zero-shot)分数的实用性,特别是在预测非编码变异效应方面。考虑到这些基于零样本的分数仅可从基因组序列中推导出来,我们鼓励将其应用于功能注释有限的非人类生物。
最后,我们已经证明通过增强模型架构可以实现降低模型规模和提高性能的双重收益。我们后续的NT-v2系列模型具有12kb的上下文长度。为了说明这一点,这个长度分别是DNABERT-1(500bp)和DNABERT-2(3kb)平均上下文长度的24倍和4倍。这些改进的模型不仅展示了更好的下游性能,而且由于其紧凑的尺寸,结合我们引入的高效微调技术,适合在经济型硬件上执行和微调,这是一个额外的优势。
虽然我们的模型仍然具有有限的注意力范围,但最近开发的Enformer模型表明,将感知域增加到200kb对于捕捉人类基因组中的长程依赖关系是必要的。作者认为,这些长程依赖关系对于准确预测基因表达是必需的,因为基因表达受远端调控元件控制,这些元件通常位于转录起始位点10-20kb以外的位置。由于自注意力机制相对于序列长度的二次方缩放,使用标准transformer架构处理如此大的输入是不可行的。Enformer模型通过在到达transformer层之前通过卷积层传递序列来减少输入维度以解决这个问题。然而,这种选择降低了其在语言建模中的有效性。另一方面,最近的HyenaDNA模型训练时的感知域达到了100万bp,但我们的下游任务基准分析表明,当训练过程中使用的感知域增加时,这些模型的性能会快速下降。
Enformer模型可参考:
基于我们的结果,我们建议开发能够处理长输入同时保持对短输入高性能的transformer模型是该领域一个有前景的方向。在多组学数据快速扩展的时代,我们最终预计本文提供的方法,以及可用的基准测试和代码,将促进基因组学中大型基础语言模型的采用、开发和改进。
方法
模型
语言模型(LM)主要在自然语言处理(NLP)领域开发,用于模拟口语。语言模型是一个在token序列(通常是词)上的概率分布,给定任何词序列,语言模型都会返回该句子存在的概率。语言模型因其能够利用大规模未标记数据集来生成通用表示而广受欢迎,这些表示即使在监督数据较少的情况下也能解决下游任务。训练语言模型的一种技术是预测序列中被遮蔽位置最可能出现的token,这通常被称为遮蔽语言建模(MLM)。
受蛋白质研究领域MLM成果的启发,该领域将蛋白质视为句子,氨基酸视为单词,我们将MLM应用于基因组学的转换器训练,将核苷酸序列视为句子,将k-mers(k=6)视为单词。转换器是一类在机器学习领域取得突破性进展的深度学习模型,包括NLP和计算机视觉。它们由一个初始嵌入层组成,该层将输入序列中的位置转换为嵌入向量,随后是一堆自注意力层,这些层依次细化这些嵌入。
用MLM训练语言模型转换器的主要技术称为双向编码器表示转换器(BERT)。在BERT中,序列中的所有位置都可以相互关注,允许信息双向流动,这在DNA序列的上下文中是必不可少的。在训练过程中,网络的最终嵌入被送入语言模型头,将其转换为输入序列上的概率分布。
架构
我们所有的模型都遵循仅编码器的转换器架构。嵌入层将token序列转换为嵌入序列。然后将位置编码添加到序列中的每个嵌入中,以向模型提供位置信息。我们使用可学习的位置编码层,最多可接受1000个token。
我们使用六聚体token作为序列长度(最多6kb)和嵌入大小之间的权衡,并且与其他token长度相比,它实现了最高的性能。然后token嵌入由转换器层堆栈处理。每个转换器层通过层归一化层和多头自注意力层转换其输入。自注意力层的输出通过跳跃连接与转换器层输入相加。该操作的结果随后通过新的层归一化层和具有GELU激活的两层感知器传递。
每个模型的头的数量、嵌入维度、感知器隐藏层中的神经元数量和层的总数可以在补充表1中找到。在自监督训练期间,堆栈最终层返回的嵌入由语言模型头转换为序列中每个位置上现有token的概率分布。
我们的第二版NT-v2模型包含一系列经证实更高效的架构变化:我们使用旋转嵌入而不是学习位置嵌入,这些嵌入在每个注意力层中使用;我们使用带有swish激活的门控线性单元,没有偏置,使NLP更有效。这些改进的模型还接受最多2048个token的序列,导致更长的上下文窗口为12kb。
训练
模型按照BERT方法进行训练。在每个训练步骤中,采样一批tokenized序列。批量大小根据可用硬件和模型大小进行调整。我们在A100 GPU集群上进行了所有实验,并采用了大小为14和2个序列的批次来训练"500M"和"2.5B"参数模型。
在序列中,15%的token子集中有80%被替换为特殊的遮蔽("MASK")token。对于人类参考基因组和多物种数据集的训练运行,15%子集中额外10%的token被替换为随机选择的标准token(任何不同于类("CLS")、填充("PAD")或MASK token 的token),就像在BERT中那样。
对于1000G数据集的训练运行,我们跳过了这种额外的数据增强,因为添加的噪声大于人类基因组中存在的自然突变频率。对于每个批次,损失函数计算为在每个选定位置上预测token概率与真实token之间的交叉熵损失之和。梯度被累积以达到每批100万个token的有效批大小。
我们使用Adam优化器,学习率计划和标准的指数衰减率和 常数,, 和 。在第一个预热期间,学习率在16,000步内在 和 之间线性增加,然后按照平方根衰减直到训练结束。
我们对NT-v2模型的超参数进行了细微调整:优化器和学习率计划保持不变;但是,我们将批量大小增加到512(每批100万个token)。受Chinchilla缩放定律的启发,与其他深度学习模型相比,我们还对NT-v2模型进行了更长时间的训练。具体来说,我们对NT-v2 50M和250M参数模型预训练了3000亿个token,而我们的"250M"和"500M"参数模型训练了高达1万亿个token,以了解相关的缩放定律。相比之下,NT-v1 2.5B参数模型训练了3000亿个token,而其500M对应模型训练了500亿个token。最后,我们使用以下NT-v2模型检查点:3000亿个token检查点用于"50M"和"100M"模型,8000亿个token检查点用于"250M"模型,以及9000亿个token检查点用于"500M"模型。
探测
我们将探测称为评估模型嵌入解决下游任务的质量。
训练后,对于每个任务,我们探测模型的每一层,并比较几种下游方法,以深入评估表示的能力。换句话说,给定下游任务的核苷酸序列数据集,我们计算并存储模型十个层返回的嵌入。然后,使用每个单独层的嵌入作为输入,我们训练了几个下游模型来解决下游任务。
我们测试了来自scikit-learn的默认超参数的逻辑回归和多层感知器(MLP)。由于我们观察到超参数的选择(如学习率、激活函数和每个隐藏层的层数)会影响最终性能,我们还对每个下游模型运行了超参数扫描。我们使用十折验证方案,其中训练数据集被分成十次训练和验证集,包含初始集的90%和10%的不同洗牌。
对于给定的超参数集,在十个分割上训练十个模型,并平均其验证性能。这个程序使用Tree-structured Parzen Estimator求解器引导超参数空间的搜索运行100次,然后在测试集上评估表现最好的模型组。因此,对于每个下游任务,对于每个预训练模型的十个层,在超参数搜索结束时记录测试集上的性能。表现最好的探测在预训练模型和其层之间的超参数在补充表8中报告。这种探测策略导致训练了76万个下游模型,这为训练和使用语言模型的各个方面提供了详细分析,例如不同层对下游任务性能的作用。
作为基线,我们评估了以tokenized序列作为输入的逻辑回归模型的性能,在通过转换器层传递token之前。使用原始tokenized序列作为输入比使用token ids被one-hot编码并通过池化层传递(在序列长度轴上求和或平均)的向量产生了更好的性能。
微调
除了通过在各个层中提取嵌入来探测我们的模型外,我们还通过IA3技术执行了参数高效的微调。使用此策略,根据任务需求将语言模型头替换为分类或回归头。
转换器层和嵌入层的权重被冻结,并引入了新的可学习权重。对于每个转换器层,我们引入了三个学习向量,和,它们在自注意力机制中被引入为:
并在位置前馈网络中被引入为 ,其中是前馈网络的非线性, 表示逐元素乘法。这总共增加了个新参数,其中是转换器层的数量。我们将这些可学习的权重称为重缩放权重。
直觉是在微调期间,这些权重将对转换器层进行加权,以改善模型在下游任务上的最终表示,使得分类/回归头能够更准确地解决问题。由于我们在探测期间观察到层专门化,我们推测这种微调技术将同样选择对特定任务具有更大预测能力的层。
实际上,重缩放权重和分类/回归头权重引入的额外参数数量约占模型总权重的0.1%。这加快了微调速度,因为只需要更新一小部分参数。同样,它也减轻了存储要求,每个下游任务只需要为500万和25亿个参数创建0.1%的新参数空间,而不是使用传统微调。例如,对于25亿参数模型,权重为9.5GB。考虑到18个下游任务,传统的微调将需要9.5×18=171GB,而参数高效的微调只需要171MB。
与探测方案类似,训练数据集被分成十次训练和验证集,包含初始集的90%和10%的不同洗牌。对于每个分割,模型微调10,000步,然后使用产生最高验证分数的参数在测试集上评估模型。我们使用批量大小为8和Adam优化器,学习率为。其他优化器参数保持与训练方案相同。每个模型对每个任务微调10,000步。这些超参数是根据NLP领域的成功经验选择的。我们的实验表明,偏离这些超参数选择并未带来显著的收益。我们还将这种方法与从随机初始化的检查点进行微调进行了比较。
与监督BPNet基线的比较
作为基线监督模型,我们从头开始在18个任务中的每一个上训练了不同变体的BPNet卷积架构。我们测试了原始架构(121,000个参数)、一个大型架构(2800万个参数)和一个特大型架构(1.13亿个参数)。对于每一个,超参数都经过手动调整以在验证集上获得最佳性能。我们实施了十折交叉验证策略来衡量每种架构的18个模型的性能。
与已发布的预训练基因组学模型的比较
我们将NT模型在18个下游任务上的微调性能与三个不同的预训练模型进行了比较:DNABERT-2、HyenaDNA(1kb和32kb上下文长度)和Enformer。我们排除了DNABERT-1,因为它最多只能处理512bp的输入长度,因此不能用于大多数任务。我们将每个模型的架构和训练权重移植到我们的代码框架中,并对每个模型的转换器部分进行如上所述的参数高效微调,使用相同的交叉验证方案进行公平比较。所有结果都可以在交互式排行榜上查看(https://huggingface.co/spaces/InstaDeepAI/nucleotide_transformer_benchmark)。只有对于HyenaDNA,由于我们的参数高效微调方法与模型架构不兼容,我们执行了完整的微调。
需要注意的是,Enformer最初是在监督方式下训练的,用于解决染色质和基因表达任务。为了进行基准测试,我们重用了提供的模型主干作为我们基准的预训练模型,这不是原始论文的预期和推荐用法;然而,我们认为这种比较很有趣,因为它突出了自监督和监督学习在预训练方面的差异,并观察到Enformer确实是一个非常有竞争力的基线,即使对于与基因表达不同的任务。不过,我们注意到Enformer最初使用不同的数据分割进行预训练,因此其在我们的基准评估中的表现可能由于潜在的数据泄露而被夸大。
预训练数据集
人类参考基因组数据集
人类参考数据集通过考虑参考组装GRCh38/hg38(https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.26)中所有常染色体和性染色体序列而构建,总计达到了32亿个核苷酸。
千人基因组(1000G)数据集
为了让模型学习人类中自然存在的遗传多样性,我们构建了一个包含来自不同人群遗传变异的训练数据集。具体来说,我们从千人基因组(1000G)计划下载了变异调用格式(VCF)文件(20201028_3202_phased),该计划旨在记录人类群体中频率至少为1%的遗传变异。
该数据集包含3,202个高覆盖度人类基因组,来自27个地理结构化的非洲、美洲、东亚和欧洲血统人群,详见附表2,总计达20.5万亿个核苷酸。这种多样性使数据集能够更好地表示人类遗传变异。为了允许从VCF文件中重建FASTA格式的单倍型,我们考虑了数据的相位版本,其中包含总计1.25亿个突变,其中1.11亿个和1400万个分别是单核苷酸多态性(SNPs)和插入缺失(indels)。
多物种数据集
多物种数据集的选择主要基于两个因素:
-
现有参考基因组的质量和
-
所使用物种间的多样性
本数据集中选择的基因组来自NCBI参考序列(RefSeq)集合(https://www.ncbi.nlm.nih.gov/)。为确保基因组集的多样性,我们从RefSeq中每个主要分组(古菌、真菌、脊椎动物、哺乳动物、脊椎动物、其他等)在属级水平上随机选择了一个基因组。然而,由于细菌基因组数量众多,我们选择只包含其随机子集。未考虑植物和病毒基因组,因为它们的调控元件与本研究关注的不同。最终筛选得到的基因组集合下采样至总计850个物种,其基因组加起来达1740亿个核苷酸。如附表3所示,数据集中每个类别在总核苷酸数中的最终贡献与从NCBI解析的原始集合中的相同。最后,我们通过选择一些在文献中被广泛研究的基因组来丰富这个数据集(附表4)。
数据准备
在收集每个基因组/个体的FASTA文件后,它们被组装成每个数据集一个唯一的FASTA文件,然后在训练前进行预处理。在数据处理阶段,所有非A、T、C、G的核苷酸都被替换为N。使用分词器将字母串转换为标记序列。分词器使用的字母表包含通过组合A、T、C、G得到的 种可能的六聚体组合,以及代表独立A、T、C、G和N的五个额外标记。它还包括三个特殊标记,即填充(PAD)、掩码(MASK)和序列开始(也称为类别(CLS))标记。这样总共构成了4,104个标记的词汇表。要对输入序列进行分词,分词器将从类别标记开始,然后从左侧开始转换序列,在可能的情况下匹配六聚体标记,或在需要时(例如出现字母N时或序列长度不是6的倍数时)回退使用独立标记。
对于多物种和人类参考数据集,基因组被分割成6,100个核苷酸的重叠片段,每个片段分别与前一个和后一个片段共享前后50个核苷酸。作为数据增强练习,对于每个训练周期和片段,在0到100之间随机采样一个起始核苷酸索引,然后从这个核苷酸开始对序列进行分词,直到达到1,000个标记。根据数据集确定训练周期数,以使模型在训练期间总共处理3000亿个标记。
在每一步,随机从训练周期集中采样一批序列输入模型。对于1000G数据集,在每一步采样人类参考基因组的序列批次(按上述方式准备)。然后,对于每个采样的片段,随机选择1000G数据集中的一个个体,如果该个体在对应于该片段的位置和染色体上携带突变,则将这些突变引入序列并替换相应的标记。这种数据处理技术确保了在训练期间对基因组和个体的均匀采样,并且使我们能够仅高效存储每个个体的突变,而不是完整的基因组。
硬件设施
所有模型都在配备有16个节点的Cambridge-1 Nvidia超级计算机系统上训练,每个节点配备8个A100 GPU,总共使用了128个A100 GPU。在训练期间,模型权重在每个GPU上复制,而批次则在GPU间分片。在每个分片上计算梯度并累积,然后在设备间平均后反向传播。我们依赖Jax库(https://jax.readthedocs.io/en/latest/_autosummary/jax.pmap.html),该库使用NCCL协议(https://developer.nvidia.com/nccl)处理节点和设备之间的通信,观察到训练时间随可用GPU数量几乎呈线性下降。500M参数模型在单个节点上训练一天,而2.5B参数模型需要整个集群训练28天。具有从5000万到5亿参数不等的NT-v2模型同样在单个节点上训练一天。所有微调运行都在配备8个A100 GPU的单个节点上执行。与训练运行一样,模型权重被复制,批次在GPU间分布。由于我们在微调时使用了8的批次大小,每个GPU在平均梯度并应用之前处理一个样本。平均而言,500M参数模型的微调运行持续20分钟,2.5B参数模型持续50分钟。
对于探测实验,所有嵌入(所有下游任务中所有序列的所有选定层)都在配备8个A100 GPU的单个节点上计算和存储,需要2天时间计算。然后,在一个由3000个CPU组成的集群上拟合76万个下游模型,需要2.5天。
Nucleotide Transformer下游任务
表观遗传标记预测
从ENCODE数据库获取了人类K562细胞系中10个组蛋白ChIP-seq数据。我们下载了以下标识符的bed narrowPeak文件:H3K4me3 (ENCFF706WUF)、H3K27ac (ENCFF544LXB)、H3K27me3 (ENCFF323WOT)、H3K4me1 (ENCFF135ZLM)、H3K36me3 (ENCFF561OUZ)、H3K9me3 (ENCFF963GZJ)、H3K9ac (ENCFF891CHI)、H3K4me2 (ENCFF749KLQ)、H4K20me1 (ENCFF909RKY)和H2AFZ (ENCFF213OTI)。对于每个数据集,我们选择包含峰值的1kb基因组序列作为正例,所有不与峰值重叠的1kb序列作为负例。
启动子序列预测
我们构建了一个启动子序列数据集来评估模型识别启动子基序的能力。我们从真核生物启动子数据库下载了所有人类启动子,跨越转录起始位点上游49bp和下游10bp(Hs_EPDnew_006_hg38.bed)。这产生了29,598个启动子区域,其中3,065个是TATA盒启动子(使用promoter_motifs.txt的基序注释)。我们选择包含启动子的300bp基因组序列作为正例,所有不与启动子重叠的300bp序列作为负例。这些正例和负例用于创建三个不同的二元分类任务:存在任何启动子元件(所有启动子)、具有TATA盒基序的启动子(TATA启动子)或不具有TATA盒基序的启动子(非TATA启动子)。
增强子序列预测
从ENCODE的SCREEN数据库获取了人类增强子元件。远端和近端增强子被合并。基于Meuleman等人的词汇表,增强子被分为组织特异性和组织不变性。与组织不变性区域重叠的增强子被定义为该类型,而所有其他增强子被定义为组织特异性。
我们选择包含增强子的400bp基因组序列作为正例,所有不与增强子重叠的400bp序列作为负例。我们创建了一个用于预测序列中是否存在增强子元件的二元分类任务(增强子)和一个多标签预测任务,标签为组织特异性增强子、组织不变性增强子或无(增强子类型)。
剪接位点预测
我们从GENCODE V44基因注释获取了所有人类注释的剪接位点。排除了level-3转录本(自动化注释)的注释,以使所有训练数据都由人工注释。我们使用HISAT2的extract_splice_sites.py来提取相应的剪接位点注释。我们选择中心包含剪接受体或供体位点的600bp基因组序列作为正例,所有不与剪接位点重叠的600bp序列作为负例。我们使用这些序列创建三个不同的任务来评估剪接预测:标签为受体、供体或无的多标签预测任务(所有剪接位点);预测剪接受体的二元分类任务(剪接受体);以及预测剪接供体的二元分类任务(剪接供体)。
数据集分割和性能评估
在人类基因组的不同染色体集上进行模型训练和性能评估。具体来说,染色体20和21的序列用于测试,其余用于训练不同的模型。删除了含有N的序列。我们通过对负例进行下采样至与正例相同数量来平衡每个数据集。为了获得可以快速基准测试任何新设计选择或模型的小型数据集,我们进一步随机对样本进行下采样,训练集最多30,000个,验证和测试集最多3,000个(详见附表5)。
我们使用十折交叉验证方案来评估每个模型,其中训练集被分成十份,每次使用其中九份进行训练,保留十分之一用于验证和选择最终检查点。我们对每个模型重复此程序十次,每次保留不同的十分之一用于验证,并在相同的保留测试集上评估最终性能。我们使用十折中的中位数性能作为每个模型在给定任务上的最终性能度量。
为了在任务间进行一致的性能评估,我们使用二元或多类MCC(Matthews相关系数)作为度量标准,因为它对不均衡的标签比例具有鲁棒性。为了最终比较模型之间的性能,我们计算了每个模型在三个不同任务类别(染色质谱系、调控元件和剪接)中的平均MCC,其中每个类别我们使用任务间的中位数MCC。
其他下游任务
染色质谱系预测
我们使用了Zhou等人编制的DeepSEA数据集进行染色质谱系预测。该数据集由240万个序列组成,每个序列大小为1000个核苷酸,并与919个染色质特征相关联。这些包括690个转录因子、125个DNase和104个组蛋白特征。如原始出版物所述,我们的模型同时在919个分类任务上进行训练,有919个独立的分类头,损失函数取为交叉熵损失的平均值。由于每个标签都高度不平衡,主要由负样本组成,因此与正样本相关的损失被放大了8倍。与DeepSEA方法不同的是,后者独立训练了两个模型,一个在正向序列上,一个在相应的反向互补序列上,并评估它们预测的平均值,而这里展示的模型仅在正向序列上训练。
SpliceAI基准测试
我们使用Illumina Basespace平台上可用的脚本来复现SpliceAI中展示的训练数据集。简而言之,这个训练数据集使用GENCODE V24lift37注释和来自GTEx队列的RNA-seq数据构建,仅关注主要转录本的剪接位点注释。
训练数据集包括位于染色体2、4、6、8和10-22以及X和Y染色体上的基因的注释,而剩余染色体上非旁系同源基因的注释构成了测试数据集。这个流程产生的每个序列长度为15,000bp,中心5,000bp包含要预测的位点。关于训练数据集构建的更多细节可在原始出版物中找到。我们调整了这个原始SpliceAI数据集以运行我们的模型,方法是将序列长度减少到6,000bp(对于NT-v1模型)和12,000bp(对于NT-v2模型),减少了侧翼上下文但保持中心5,000bp。我们还删除了包含N的序列。在与第一个数据集上的SpliceAI进行比较时(我们称之为SpliceAI-6k),我们附加了9,000个"N"核苷酸作为侧翼序列,因为SpliceAI基于具有15,000bp输入的模型。在与第二个数据集上的SpliceAI进行比较时,我们报告了原始出版物中呈现的性能,与本数据集相比,包括15,000bp而不是12,000bp的序列长度。
这个任务是一个类似于SpliceAI的多标签分类任务,针对输入序列的每个核苷酸(图5d)。对于transformer模型输出的每个嵌入,一个头部为该标记嵌入所代表的六个核苷酸中的每一个预测三个标签概率:剪接受体、剪接供体或无。该头部是一个简单的分类层,预测18个类别(六个核苷酸中的每一个都有三个标签)。为确保每个嵌入都与一个六聚体相关联,序列被切割以使其长度能被6整除。
此外,从训练和测试集中删除了所有包含N的序列,这仅占数据的很少一部分。值得注意的是,如果我们使用字节对编码分词器(如DNABERT-2),每个嵌入所代表的核苷酸数量会变化,这会使核苷酸级别的预测任务变得更加困难。
增强子活性预测
我们使用了de Almeida等人发布的DeepSTARR增强子活性数据集。该数据集由484,052个大小为249个核苷酸的DNA序列组成,每个序列都测量了它们对发育型或管家型启动子的定量增强子活性。
我们在模型中添加了两个独立的回归头以同时预测这两种增强子活性。遵循先前工作的方法,我们选择将这个回归任务作为多标签分类问题处理。
具体来说,每个标签被离散化为一组50个值 ,在最小值和最大值之间均匀分布。对于每个标签,模型预测归一化权重 ,使得:
其他性能分析
t-SNE嵌入投影
使用 t-分布随机邻域嵌入(t-SNE)将NT内部嵌入降维到二维向量,以可视化不同基因组元件的分离情况。在几个transformer层输出计算了每个基因组元件的NT嵌入,并计算对应于该元件的序列位置的平均嵌入。然后将这些平均嵌入作为输入传递到带有默认参数的sklearn Python包中的t-SNE降维对象。
重构准确率和复杂度(perplexity)
我们研究了预训练模型如何重构被掩码的标记。我们考虑一个带参数 的训练好的语言模型。在一个感兴趣的核苷酸序列 内,我们使用两种策略之一来掩码标记(我们用掩码(MASK)标记替换这些位置的标记)。
我们要么只掩码序列的中心标记,要么随机掩码序列内15%的标记。然后将掩码序列输入模型,并检索每个掩码位置的标记概率。损失函数 和准确率 定义如下:
其中 是掩码位置的集合, 是词汇表(所有现有标记的集合)。
复杂度通常在自回归生成模型的上下文中定义。这里,我们依赖Rives提出的另一种定义,将其定义为在掩码位置上计算的损失函数的指数:
复杂度衡量模型重构掩码位置的能力,比准确率更精细,因为它也考虑了幅度。与准确率相反,较低的复杂度表示更好的重构能力,因此性能更好。
不同基因组元素中的标记重构
我们还在整个22号染色体上对 6kb 窗口执行了这种标记重构方法。我们只保留没有N的窗口。对于每个窗口,每次掩码一个标记,恢复序列中原始标记的预测概率。
我们在WashU表观基因组浏览器会话中显示这些分数(https://shorturl.at/jov28)。为了获得不同类型基因组元素的平均标记概率,我们从Ensembl(外显子、内含子、剪接受体和供体、5’UTR、3’UTR、转录起始位点和转录终止位点)(https://www.ensembl.org/info/data/ftp/index.html)、GENCODE的polyA信号位点(https://www.gencodegenes.org/human/)和ENCODE的调控元件(来自SCREEN数据库的增强子、启动子和CTCF结合位点)(https://api.wenglab.org/screen_v13/fdownloads/GRCh38-ccREs.bed)检索基因注释区域。
功能变异优先级排序
为了获得具有不同严重程度的遗传变异,我们使用了变异效应预测(VEP)软件并对整个人类基因组的序列进行注释。
具体来说,我们在整个人类基因组中随机采样序列,并保留了注释为以下任何类别的序列中的遗传变异:"内含子变异"、"基因间变异"、"调控区域变异"、"错义变异"、"3'UTR变异"、"同义变异"、"TF结合位点变异"、"5'UTR变异"、"剪接区域变异"和"终止获得变异"。
在只保留SNP并过滤掉注释为多个结果的变异(例如那些注释为终止获得和剪接变异的变异)后,我们获得了每个类别920个遗传变异的最终数据集。
作为功能性遗传变异的正集,我们从四个不同来源编制了SNP。我们使用来自GRASP数据库的与基因表达(eQTL)和meQTL相关的SNP, 值 <,来自ClinVar的带有"可能致病"注释的SNP以及HGMD(公共v.2020.4)中报告的SNP。
经过这些过滤,我们分别保留了eQTL、meQTL、ClinVar和HGMD SNP数据集的总计80,590、11,734、70,224和14,626个遗传变异。对于这四个数据集中的每一个,我们然后基于来自1000G项目的SNP构建了一组阴性变异,这些SNP的次要等位基因频率>5%,与测试数据集中报告的任何变异都不重叠,并且在相关变异的100 kb内,从而产生四个平衡数据集。
对于给定的感兴趣位点,要计算基于零样本的分数,我们执行以下操作:对于每个SNP,我们基于人类参考基因组获得以SNP为中心的6,000 bp序列。然后我们创建两个序列,一个携带SNP位置的参考等位基因,另一个携带替代等位基因。然后我们计算几个零样本分数,这些分数捕获这两个序列之间嵌入空间中的矢量距离的不同方面,即:
-
L1距离(曼哈顿距离)
-
L2距离(欧几里得距离)
-
余弦相似度
-
点积(未归一化的余弦相似度)
我们还计算了替代等位基因的损失以及携带替代和参考等位基因的序列之间的损失差异,作为另外两个零样本分数。在功能性变异的情况下,除了零样本分数外,我们还微调了transformer模型来分类阳性和阴性变异。我们采用了类似于先前描述的策略,主要区别在于训练集和测试集按染色体划分,并严格保持不重叠。
具体来说,我们将22个染色体分为五组,依次使用每组作为测试集,其他四组作为训练集。通过微调训练集,我们可以得出测试集中每个序列作为阳性变异的概率。我们使用这些概率作为每个SNP的分数。
为了将这些预测与其他方法进行比较,我们从四个数据集中的每一个随机抽样10,000个阳性和阴性SNP。然后我们使用组合注释依赖性消减工具(v.GRCh38-v1.6)来计算CADD、GERP、phastCons和phyloP分数。DeepSEA分数是使用 https://hb.flatironinstitute.org/sei/ 提供的Beluga模型计算的。考虑的分数是报告给每个SNP的"疾病影响分数"。
注意力图分析
我们分析了从预训练模型收集的注意力图如何捕获关键基因组元素。我们遵循先前工作中提出的方法。对于基因组元素,我们定义了标记上的指示函数 ,如果标记 内的一个或多个核苷酸属于该元素,则等于1,否则等于0。
我们计算了在一个注意力头中,在核苷酸序列数据集 上聚合的、集中在该基因组元素上的注意力的平均比例:
其中 是标记 和标记 之间的注意力系数,定义为 , 是置信度阈值。
我们计算了所有模型的所有头和所有层的 值,并考虑了九个元素("5'UTR"、"3'UTR"、"外显子"、"内含子"、"增强子"、"启动子"、"CTCF结合位点"、"开放染色质"和"转录因子结合位点")。
我们在一个由90,000个序列组成的数据集上执行这些分析,每个特征10,000个,长度为6 kb,从人类参考基因组中提取。每个元素所属标记的平均比例可以在补充表10中找到。对于每个序列,在数据集创建期间均匀采样特征在序列中的位置。如先前工作所建议,我们为所有实验选择了置信度阈值 。
如果 的数量显著大于该特征在数据集中的自然出现频率(补充表10),我们认为该特征被注意力头捕获。为了验证这一点,我们进行了两个比例的z检验,零假设为特征的自然频率,备择假设为 。使用每个模型的总头数作为 0.05 显著性水平 的Bonferroni校正。我们为每个模型中的每个头部对每个基因组元素计算z分数和相关的P值,如下所示:
其中 表示与每个基因组元素相关的高于 的注意力的比例, 表示序列中被该基因组元素占据的比例, 是注意力高于 的序列位置总数, 是序列位置总数。P值低于Bonferroni校正显著性水平的注意力头被认为是显著的。
预测DeepSTARR数据中重要的TF基序实例
我们从DeepSTARR数据集中检索实验突变数据,其中单个TF基序实例被突变,并测量它们对发育和管家增强子活性的影响。我们评估了完全微调的NT 2.5B Multispecies模型(因为它是测试集性能最高的模型)通过预测野生型和相应基序突变序列的活性并计算它们的 倍数变化来预测每个TF基序实例贡献的性能。我们将我们预测的突变效应与原始方法DeepSTARR预测的效应和实验得到的 倍数变化进行了比较。
数据可用性
NT预训练序列来自公开可用的资源。1000G序列从1000G项目数据库获得,人类和多物种参考基因组从NCBI RefSeq数据库获得。基因注释从GENCODE和Ensembl数据库获得。使用Ensembl的VEP API获得变异效应预测。致病性和调控变异从ClinVar、GRASP和HGMD(公共v.2020.4)通过Ensembl Biomart提取。NT下游任务是从以下公开可用资源中整理的:ENCODE、SCREEN和DHSIndex、真核生物启动子数据库和GENCODE(补充表5)。我们在HuggingFace上提供了我们的多物种和人类预训练数据集版本(NT models集合)以及我们的下游任务基准测试(NT下游任务集)。我们建立了一个交互式排行榜,其中包含所有模型在每个任务上的结果,以便于比较。我们还在WashU表观基因组浏览器上创建了一个交互式浏览器会话,显示整个22号染色体上预训练模型的标记概率。
代码可用性
预训练transformer模型的代码和权重以及Jax中的推理代码可在NT GitHub仓库获得用于研究目的。PyTorch中模型的HuggingFace版本可在NT HuggingFace集合找到。示例笔记本可在HuggingFace的PyTorch生物示例页面获得。
补充信息
补充说明,补充表1-12和补充图1-20。请在这里下载。
参考文献
-
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. Bert: pre-training of deep bidirectional transformers for language understanding. Preprint at https://arxiv.org/abs/1810.04805 (2018).
-
Brown, T. et al. Language models are few-shot learners. Adv. Neural Inf. Process. (2020).
-
Jumper, J. et al. Highly accurate protein structure prediction with alphafold. Nature 596, 583-589 (2021).
-
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA 118, e2016239118 (2021).
-
Elnaggar, A. et al. ProtTrans: towards cracking the language of life’s code through self-supervised deep learning and high performance computing. IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112-7127 (2022).
-
Littmann, M., Heinzinger, M., Dallago, C., Olenyi, T. & Rost, B. Embeddings from deep learning transfer go annotations beyond homology. Sci. Rep. 11, 1-14 (2021).
-
Marquet, C. et al. Embeddings from protein language models predict conservation and variant effects. Hum. Genet. 141, 1629-1647 (2022).
-
Littmann, M., Heinzinger, M., Dallago, C., Weissenow, K. & Rost, B. Protein embeddings and deep learning predict binding residues for various ligand classes. Sci. Rep. 11, 23916 (2021).
-
Avsec, Ž. et al. Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet. 53, 354-366 (2021).
-
Zhou, J. & Troyanskaya, O. G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 12, 931-934 (2015).
-
Mateo, L. J., Sinnott-Armstrong, N. & Boettiger, A. N. Tracing dna paths and rna profiles in cultured cells and tissues with orca. Nat. Protoc. 16, 1647-1713 (2021).
-
de Almeida, B. P., Reiter, F., Pagani, M. & Stark, A. DeepSTARR predicts enhancer activity from dna sequence and enables the de novo design of synthetic enhancers. Nat. Genet. 54, 613-624 (2022).
-
Eraslan, G., Avsec, Ž., Gagneur, J. & Theis, F. J. Deep learning: new computational modelling techniques for genomics. Nat. Rev. Genet. 20, 389-403 (2019).
-
Zhou, J. et al. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. 50, 1171-1179 (2018).
-
Kelley, D. R. Cross-species regulatory sequence activity prediction. PLOS Comput. Biol. 16, e1008050 (2020).
-
Kelley, D. R. et al. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res. 28, 739-750 (2018).
-
Agarwal, V. & Shendure, J. Predicting mRNA abundance directly from genomic sequence using deep convolutional neural networks. Cell Rep. 31, 107663 (2020).
-
Chen, K. M., Wong, A. K., Troyanskaya, O. G. & Zhou, J. A sequence-based global map of regulatory activity for deciphering human genetics. Nat. Genet. 54, 940-949 (2022).
-
Avsec, Ž. et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 18, 1196-1203 (2021).
-
Ji, Y., Zhou, Z., Liu, H. & Davuluri, R. V. Dnabert: pre-trained bidirectional encoder representations from transformers model for DNA-language in genome. Bioinformatics 37, 2112-2120 (2021).
-
Zvyagin, M. T. et al. Genslms: genome-scale language models reveal SARS-CoV-2 evolutionary dynamics. Preprint at bioRXiv https://doi.org/10.1101/2022.10.10.511571 (2022).
-
Oute, J. C. & Deane, C. M. Protein language embeddings provide strong signals for protein engineering. Nat. Mach. Intell. 6, 170-179 (2024).
-
Zhou, Z. et al. Dnabert-2: efficient foundation model and benchmark for multi-species genome. in Proceedings of the Twelfth International Conference on Learning Representations https://openreview.net/pdf?id=oMLQB4EZE1 (ICLR, 2024).
-
Fishman, V. et al. Gena-lm: A family of open-source foundational models for long dna sequences. Preprint at bioRXiv https://doi.org/10.1101/2023.06.12.544594 (2023).
-
Nguyen, E. et al. Hyenadna: Long-range genomic sequence modeling at single nucleotide resolution. in 37th Conference on Neural Information Processing Systems https://openreview.net/pdf?id=ubzNoJjOKj (NeurIPS, 2023).
-
Mendoza-Revilla, J. et al. A foundational large language model for edible plant genomes. Commun. Biol. 7, 835 (2024).
-
Rae, J. W. et al. Scaling language models: methods, analysis & insights from training gopher. Preprint at https://arxiv.org/abs/2112.11446 (2021).
-
Consortium, G. P. et al. A global reference for human genetic variation. Nature 526, 68 (2015).
-
Harrow, J. et al. GENCODE: the reference human genome annotation for the encode project. Genome Res. 22, 1760-1774 (2012).
-
Meylan, P., Dreos, R., Ambrosini, G., Groux, R. & Bucher, P. Epd in 2020: enhanced data visualization and extension to ncRNA promoters. Nucleic Acids Res. 48, D65-D69 (2020).
-
ENCODE. An integrated encyclopedia of dna elements in the human genome. Nature 489, 57-74 (2012).
-
The ENCODE Project Consortium. Expanded encyclopaedias of dna elements in the human and mouse genomes. Nature 583, 699-710 (2020).
-
Meuleman, W. et al. Index and biological spectrum of human DNase I hypersensitive sites. Nature 584, 244-251 (2020).
-
Li, F.-Z., Amini, A. P., Yang, K. K. & Lu, A. X. Pretrained protein language model transfer learning: is the final layer representation what we want? in Machine Learning for Structural Biology Workshop (NeurIPS, 2022).
-
Liu, H. et al. Few-shot parameter-efficient fine-tuning is better and cheaper than in-context learning. in 36th Conference on Neural Information Processing Systems (NeurIPS, 2022).
-
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535-548 (2019).
-
Benegas, G., Batra, S. S. & Song, Y. S. DNA language models are powerful zero-shot predictors of non-coding variant effects. Proc. Natl Acad. Sci. USA 120, e2311219120 (2023).
-
Vig, J. et al. BERTology meets biology: Interpreting attention in protein language models. in Proceedings of the International Conference on Learning Representations 2021 https://openreview.net/pdf?id=YWtLZvLmud7 (ICLR, 2021).
-
Braun, S. et al. Decoding a cancer-relevant splicing decision in the ron proto-oncogene using high-throughput mutagenesis. Nat. Commun. 9, 3315 (2018).
-
McLaren, W. et al. The Ensembl variant effect predictor. Genome Biol. 17, 1-14 (2016).
-
Lappalainen, T. et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506-511 (2013).
-
Consortium, G. The gtex consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318-1330 (2020).
-
Võsa, U. et al. Large-scale cis-and trans-eqtl analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genetics 53, 1300-1310 (2021).
-
Chowdhery, A. et al. PaLM: scaling language modeling with pathways. J. Mach. Learn. Technol. 24, 11324-11436 (2021).
-
Hoffmann, J. et al. Training compute-optimal large language models. in 36th Conference on Neural Information Processing Systems https://proceedings.neurips.cc/paper_files/paper/2022/file/c1e2faff6f58887093f114ebe04a3e5-Paper-Conference.pdf (NeurIPS, 2022).
-
Rogers, A., Kovaleva, O. & Rumshisky, A. A primer in BERTology: what we know about how bert works. Trans. Assoc. Comput. Linguist. 8, 842-866 (2020).
-
Stärk, H., Dallago, C., Heinzinger, M. & Rost, B. Light attention predicts protein location from the language of life. Bioinform. Adv. 1, vbab035 (2021).
-
Zou, J. et al. A primer on deep learning in genomics. Nat. Genetics 51, 12-18 (2019).
-
Wang, A. et al. Superglue: a stickier benchmark for general-purpose language understanding systems. in 33rd Conference on Neural Information Processing Systems https://papers.nips.cc/paper_files/paper/2019/file/4496bf24afe7fab6f046bf4923da8de6-Paper.pdf (NeurIPS, 2019).
-
Hendrycks, D. & Gimpel, K. Gaussian error linear units (gelus). Preprint at https://arxiv.org/abs/1606.08415 (2016).
-
Su, J. et al. Roformer: enhanced transformer with rotary position embedding. Neurocomputing 568, 127063 (2024).
-
Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization. Preprint at https://arxiv.org/abs/1412.6980v5 (2015).
-
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825-2830 (2011).
-
Bergstra, J., Bardenet, R., Bengio, Y. & Kégl, B. Algorithms for hyper-parameter optimization. in Advances in Neural Information Processing Systems 24 https://papers.nips.cc/paper_files/paper/2011/file/86e8f7ab32cfd12577bc2619bc635690-Paper.pdf (NeurIPS, 2011).
-
Byrska-Bishop, M. et al. High-coverage whole-genome sequencing of the expanded 1000 genomes project cohort including 602 trios. Cell 185, 3426-3440 (2022).
-
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with hisat2 and hisat-genotype. Nat. Biotechnol. 37, 907-905 (2019).
-
Leslie, R., O’Donnell, C. J. & Johnson, A. D. GRASP: analysis of genotype-phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics 30, i185-i194 (2014).
-
Landrum, M. J. et al. Clinvar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062-D1067 (2018).
-
Stenson, P. D. et al. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 139, 1197-1207 (2020).
